Ullah Inayat, Kabir Firoz, Iqbal Muhammad, Gottsch Clare Brooks S, Naeem Muhammad Asif, Assir Muhammad Zaman, Khan Shaheen N, Akram Javed, Riazuddin Sheikh, Ayyagari Radha, Hejtmancik J Fielding, Riazuddin S Amer
National Centre of Excellence in Molecular Biology, University of the Punjab, Lahore, Pakistan.
The Wilmer Eye Institute, Johns Hopkins University School of Medicine, Baltimore, MD.
Mol Vis. 2016 Jul 16;22:797-815. eCollection 2016.
To identify pathogenic mutations responsible for autosomal recessive retinitis pigmentosa (arRP) in consanguineous familial cases.
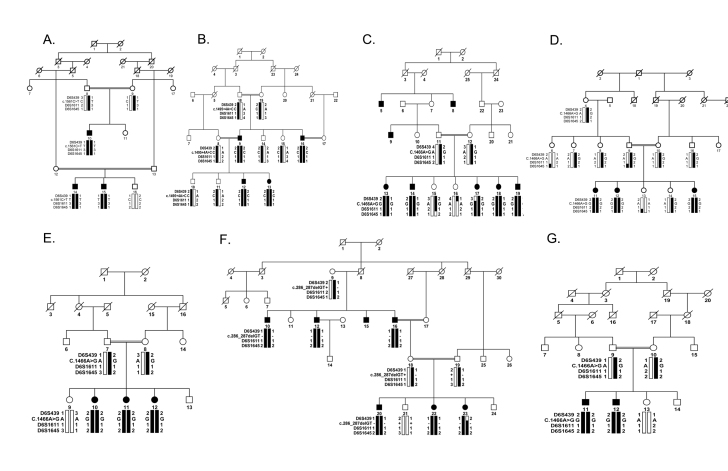
Seven large familial cases with multiple individuals diagnosed with retinitis pigmentosa were included in the study. Affected individuals in these families underwent ophthalmic examinations to document the symptoms and confirm the initial diagnosis. Blood samples were collected from all participating members, and genomic DNA was extracted. An exclusion analysis with microsatellite markers spanning the TULP1 locus on chromosome 6p was performed, and two-point logarithm of odds (LOD) scores were calculated. All coding exons along with the exon-intron boundaries of TULP1 were sequenced bidirectionally. We constructed a single nucleotide polymorphism (SNP) haplotype for the four familial cases harboring the K489R allele and estimated the likelihood of a founder effect.
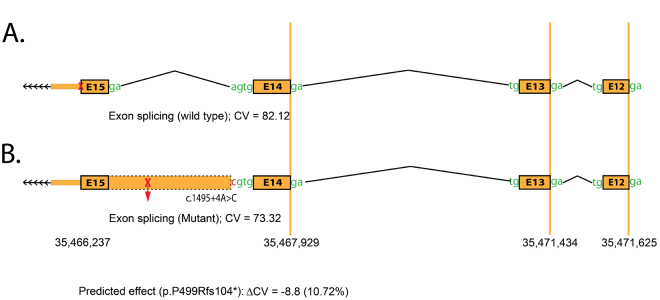
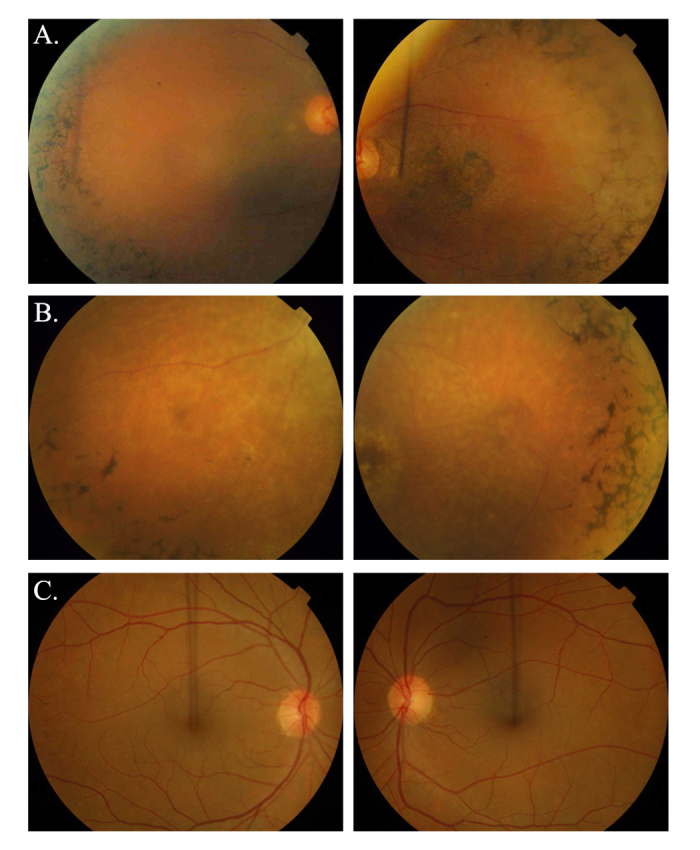
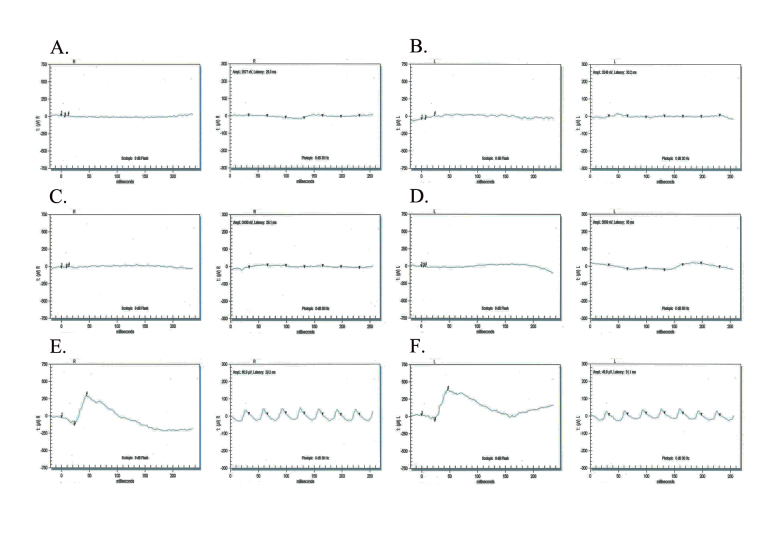
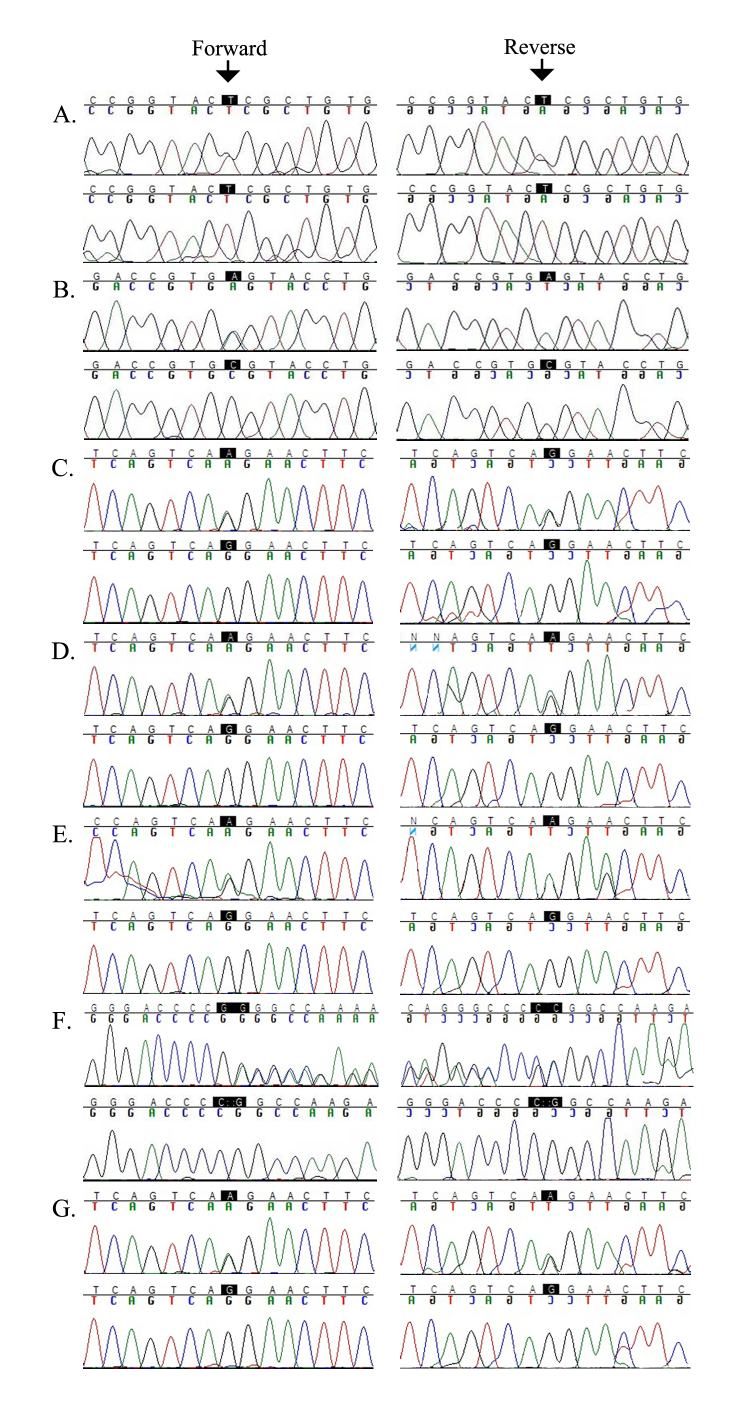
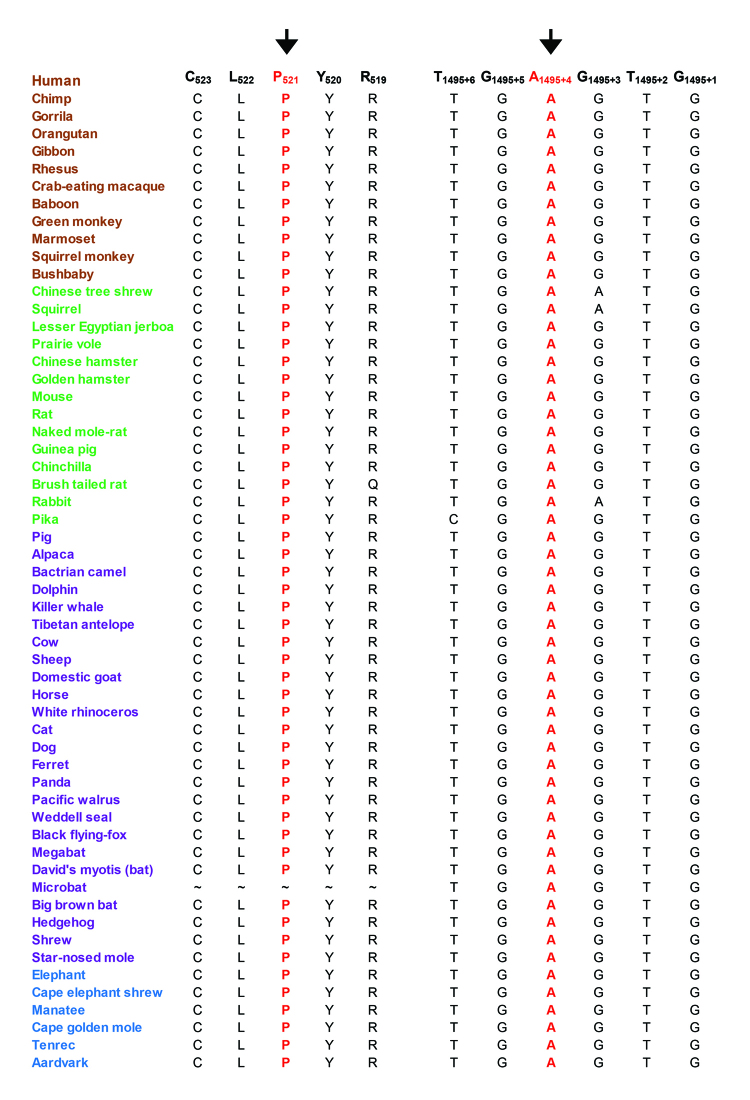
The ophthalmic examinations of the affected individuals in these familial cases were suggestive of RP. Exclusion analyses confirmed linkage to chromosome 6p harboring TULP1 with positive two-point LOD scores. Subsequent Sanger sequencing identified the single base pair substitution in exon14, c.1466A>G (p.K489R), in four families. Additionally, we identified a two-base deletion in exon 4, c.286_287delGA (p.E96Gfs77*); a homozygous splice site variant in intron 14, c.1495+4A>C; and a novel missense variation in exon 15, c.1561C>T (p.P521S). All mutations segregated with the disease phenotype in the respective families and were absent in ethnically matched control chromosomes. Haplotype analysis suggested (p<10(-6)) that affected individuals inherited the causal mutation from a common ancestor.
Pathogenic mutations in TULP1 are responsible for the RP phenotype in seven familial cases with a common ancestral mutation responsible for the disease phenotype in four of the seven families.
在近亲家族性病例中鉴定导致常染色体隐性遗传性视网膜色素变性(arRP)的致病突变。
本研究纳入了7个有多名个体被诊断为视网膜色素变性的大型家族性病例。这些家族中的患病个体接受了眼科检查,以记录症状并确认初步诊断。从所有参与成员中采集血样,提取基因组DNA。使用跨越6号染色体p臂上TULP1基因座的微卫星标记进行排除分析,并计算两点对数优势(LOD)分数。对TULP1的所有编码外显子以及外显子-内含子边界进行双向测序。我们为携带K489R等位基因的4个家族性病例构建了单核苷酸多态性(SNP)单倍型,并估计了奠基者效应的可能性。
这些家族性病例中患病个体的眼科检查提示为RP。排除分析证实与含有TULP1的6号染色体p臂连锁,两点LOD分数为阳性。随后的桑格测序在4个家族中鉴定出第14外显子中的单碱基对替换,即c.1466A>G(p.K489R)。此外,我们在第4外显子中鉴定出一个两碱基缺失,即c.286_287delGA(p.E96Gfs77*);在第14内含子中鉴定出一个纯合剪接位点变异,即c.1495+4A>C;以及在第15外显子中鉴定出一个新的错义变异,即c.1561C>T(p.P521S)。所有突变在各自家族中均与疾病表型共分离,并且在种族匹配的对照染色体中不存在。单倍型分析表明(p<10(-6)),患病个体从共同祖先那里继承了致病突变。
TULP1中的致病突变导致了7个家族性病例中的RP表型,其中7个家族中有4个家族的疾病表型由一个共同的祖先突变引起。