Song Hongxin, Rossi Ethan A, Stone Edwin, Latchney Lisa, Williams David, Dubra Alfredo, Chung Mina
Beijing Institute of Ophthalmology, Beijing Tongren Eye Center, Beijing Tongren Hospital, Capital Medical University, Beijing Key Laboratory of Ophthalmology and Visual SciencesNational Engineering Research Center for Ophthalmic Equipment, Beijing, China.
University of Rochester, Center for Visual Science, Rochester, New York, USA.
Br J Ophthalmol. 2018 Jan;102(1):136-141. doi: 10.1136/bjophthalmol-2017-310498. Epub 2017 Oct 26.
Several genes causing autosomal-dominant cone-rod dystrophy (AD-CRD) have been identified. However, the mechanisms by which genetic mutations lead to cellular loss in human disease remain poorly understood. Here we combine genotyping with high-resolution adaptive optics retinal imaging to elucidate the retinal phenotype at a cellular level in patients with AD-CRD harbouring a defect in the gene.
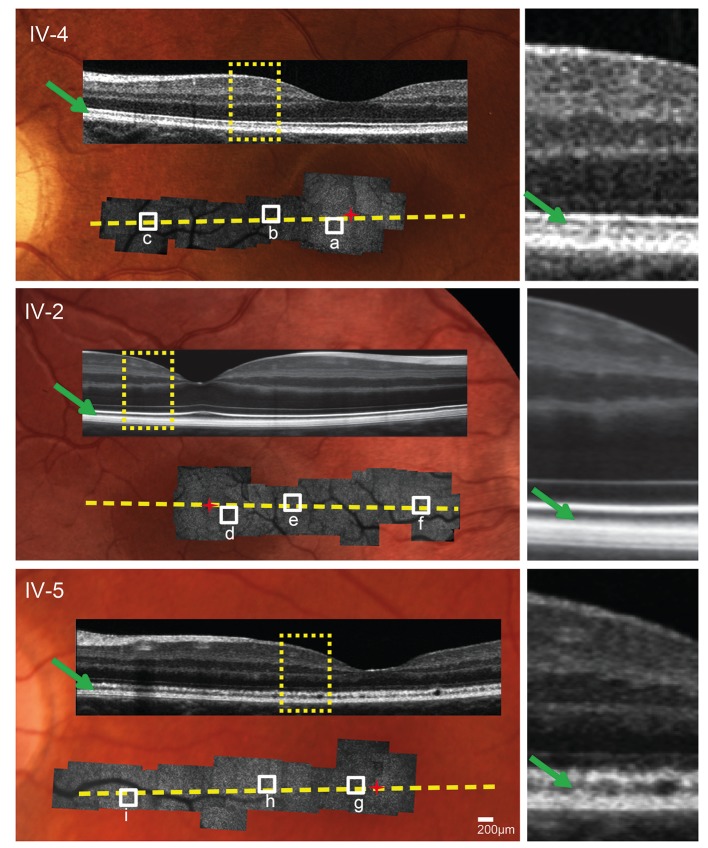
Nine affected members of a four-generation AD-CRD pedigree and three unaffected first-degree relatives underwent clinical examinations including visual acuity, fundus examination, Goldmann perimetry, spectral domain optical coherence tomography and electroretinography. Genome-wide scan followed by bidirectional sequencing was performed on all affected participants. High-resolution imaging using a custom adaptive optics scanning light ophthalmoscope (AOSLO) was performed for selected participants.
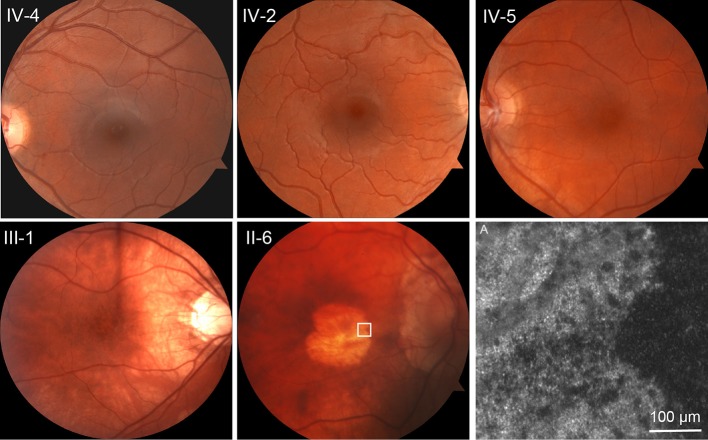
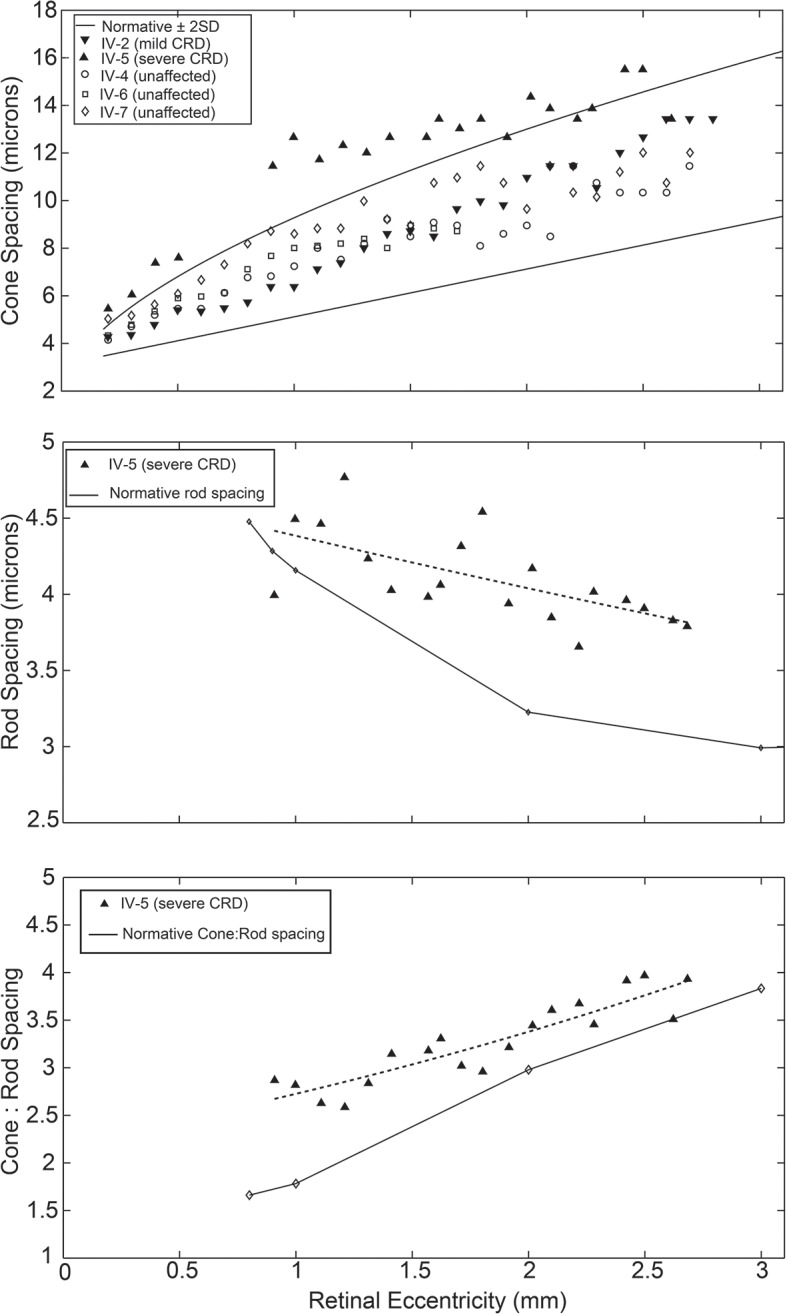
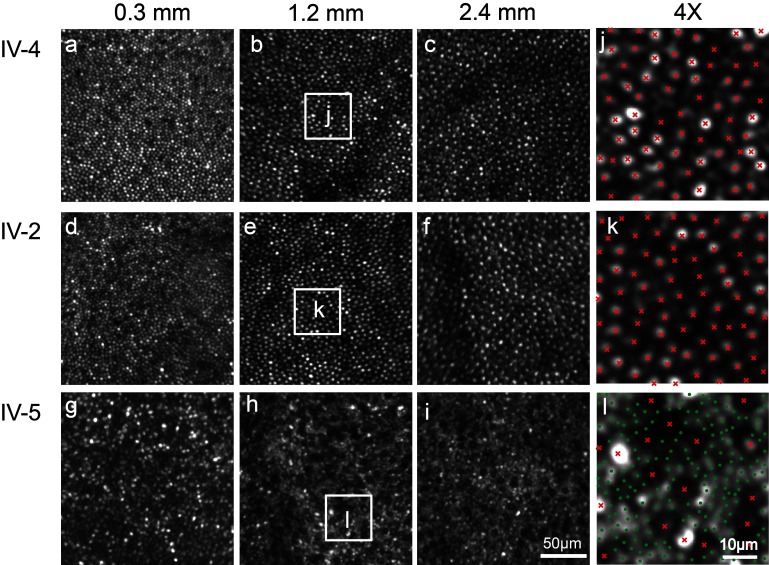
Clinical evaluations showed a range of disease severity from normal fundus appearance in teenaged patients to pronounced macular atrophy in older patients. Molecular genetic testing showed a mutation in in segregating with disease. AOSLO imaging revealed that of the two teenage patients with mild disease, one had severe disruption of the photoreceptor mosaic while the other had a normal cone mosaic.
AOSLO imaging demonstrated variability in the pattern of cone and rod cell loss between two teenage cousins with early AD-CRD, who had similar clinical features and had the identical disease-causing mutation in . This finding suggests that a mutation in does not lead to the same degree of AD-CRD in all patients. Modifying factors may mitigate or augment disease severity, leading to different retinal cellular phenotypes.
已鉴定出几种导致常染色体显性遗传性锥杆营养不良(AD-CRD)的基因。然而,基因突变导致人类疾病中细胞丢失的机制仍知之甚少。在此,我们将基因分型与高分辨率自适应光学视网膜成像相结合,以在细胞水平上阐明携带该基因缺陷的AD-CRD患者的视网膜表型。
对一个四代AD-CRD家系的9名患病成员和3名未患病的一级亲属进行了临床检查,包括视力、眼底检查、Goldmann视野检查、光谱域光学相干断层扫描和视网膜电图检查。对所有患病参与者进行全基因组扫描,随后进行双向测序。对选定的参与者使用定制的自适应光学扫描激光检眼镜(AOSLO)进行高分辨率成像。
临床评估显示疾病严重程度范围较广,从青少年患者眼底外观正常到老年患者明显的黄斑萎缩。分子遗传学检测显示该基因存在与疾病共分离的突变。AOSLO成像显示,在两名患有轻度疾病的青少年患者中,一名患者的光感受器镶嵌严重破坏,而另一名患者的视锥镶嵌正常。
AOSLO成像显示,两名患有早期AD-CRD的青少年表亲,尽管临床特征相似且致病突变相同,但视锥和视杆细胞丢失模式存在差异。这一发现表明,该基因突变在所有患者中不会导致相同程度的AD-CRD。修饰因子可能会减轻或加重疾病严重程度,导致不同的视网膜细胞表型。