Cambridge Institute for Medical Research, University of Cambridge, Wellcome Trust/MRC Building, Cambridge CB2 0XY, England.
Acta Crystallogr D Struct Biol. 2018 Jun 1;74(Pt 6):519-530. doi: 10.1107/S2059798318002425. Epub 2018 Apr 11.
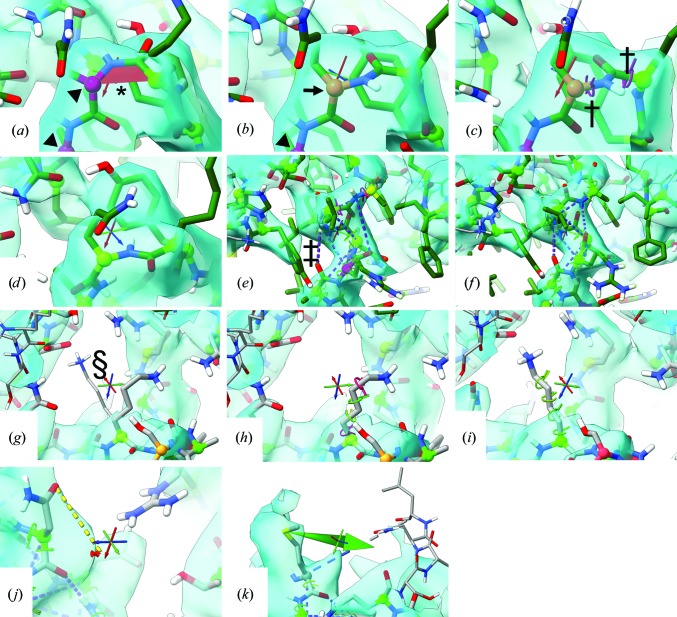
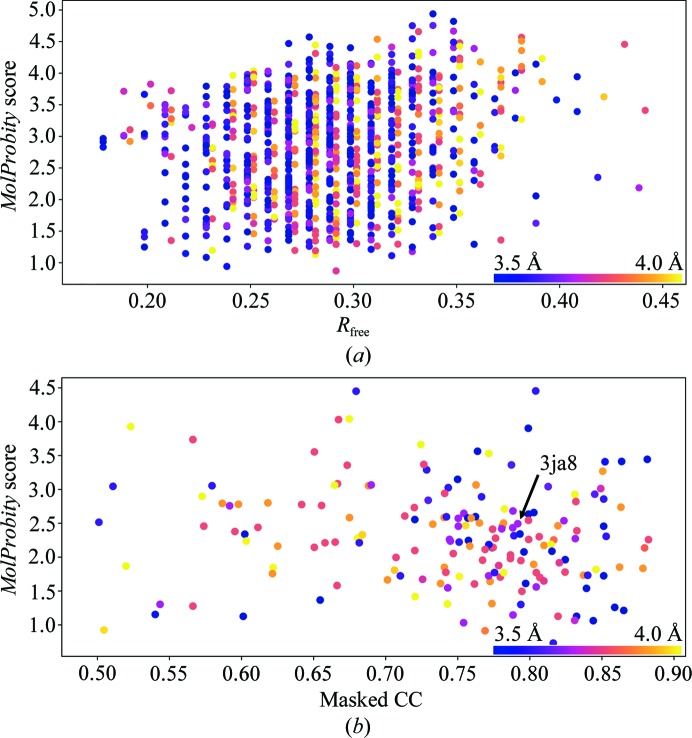
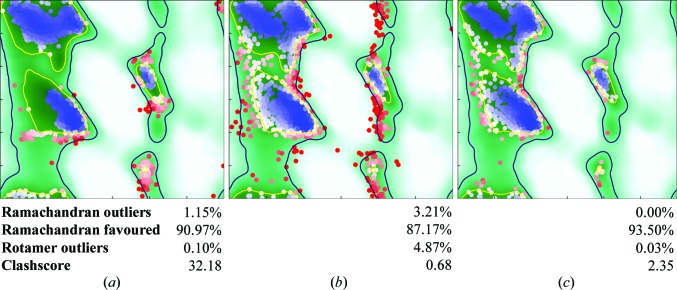
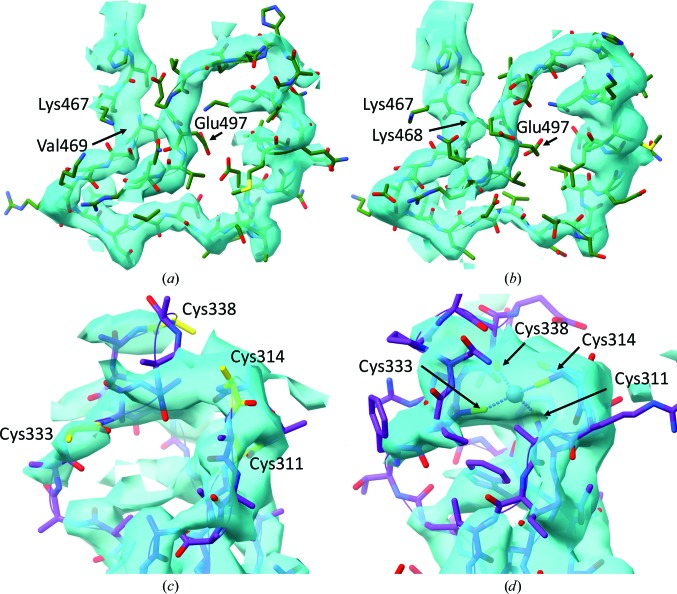
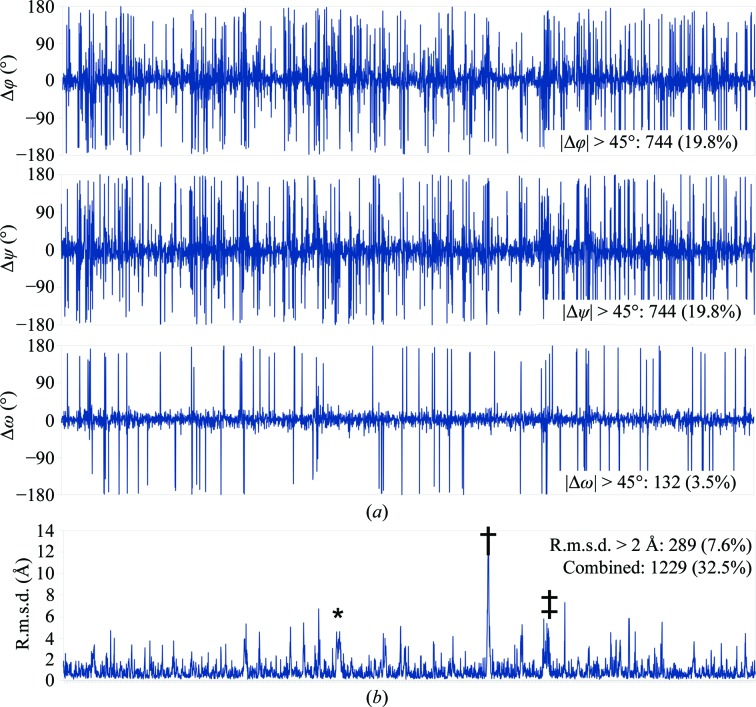
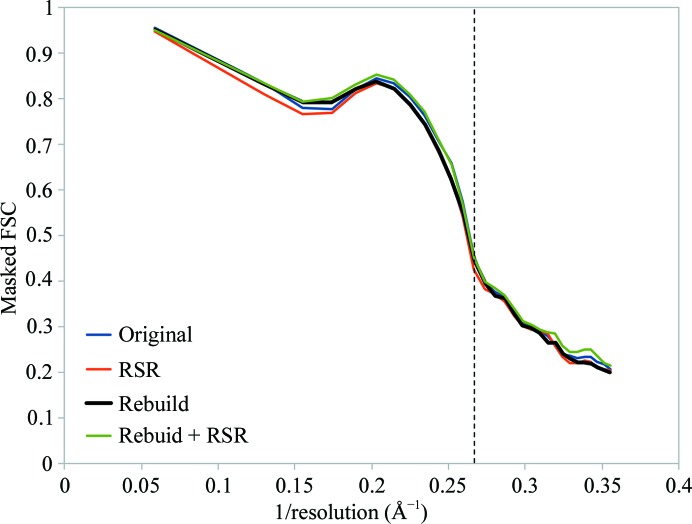
This paper introduces ISOLDE, a new software package designed to provide an intuitive environment for high-fidelity interactive remodelling/refinement of macromolecular models into electron-density maps. ISOLDE combines interactive molecular-dynamics flexible fitting with modern molecular-graphics visualization and established structural biology libraries to provide an immersive interface wherein the model constantly acts to maintain physically realistic conformations as the user interacts with it by directly tugging atoms with a mouse or haptic interface or applying/removing restraints. In addition, common validation tasks are accelerated and visualized in real time. Using the recently described 3.8 Å resolution cryo-EM structure of the eukaryotic minichromosome maintenance (MCM) helicase complex as a case study, it is demonstrated how ISOLDE can be used alongside other modern refinement tools to avoid common pitfalls of low-resolution modelling and improve the quality of the final model. A detailed analysis of changes between the initial and final model provides a somewhat sobering insight into the dangers of relying on a small number of validation metrics to judge the quality of a low-resolution model.
本文介绍了 ISOLDE,这是一个新的软件包,旨在为高保真交互式重塑/细化大分子模型成电子密度图提供直观的环境。ISOLDE 将交互式分子动力学灵活拟合与现代分子图形可视化和成熟的结构生物学库相结合,提供了一个沉浸式接口,在该接口中,模型通过直接用鼠标或触觉界面拖拉原子或应用/移除约束,不断作用以保持物理上逼真的构象,而用户与之交互。此外,常见的验证任务也被加速并实时可视化。本文以最近描述的分辨率为 3.8 Å 的真核微小染色体维持(MCM)解旋酶复合物的冷冻电镜结构为例,展示了如何与其他现代精修工具一起使用 ISOLDE,以避免低分辨率建模的常见陷阱并提高最终模型的质量。对初始模型和最终模型之间变化的详细分析,对仅依赖少数验证指标来判断低分辨率模型质量的危险,提供了一个清醒的认识。