Chair of Proteomics and Bioanalytics, Technische Universität München, Freising, Germany; Bavarian Biomolecular Mass Spectrometry Center (BayBioMS), TUM, Freising, Germany.
Department of Data Science, Division of Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215.
Mol Cell Proteomics. 2019 Aug 9;18(8 suppl 1):S153-S168. doi: 10.1074/mcp.TIR118.001251. Epub 2019 Jun 26.
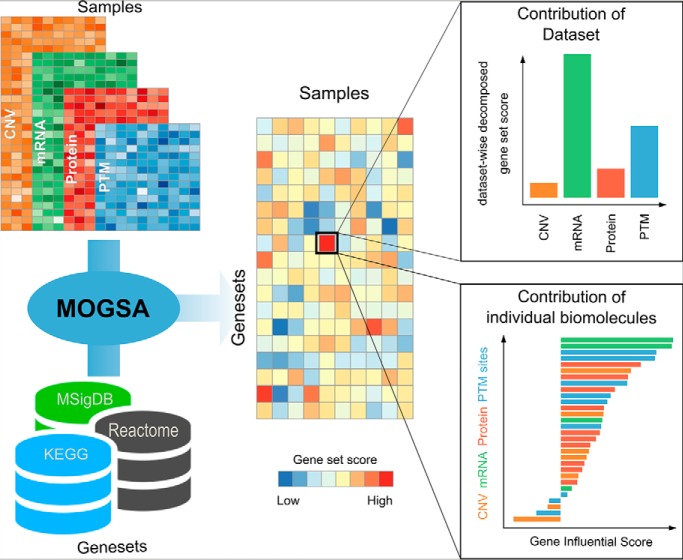
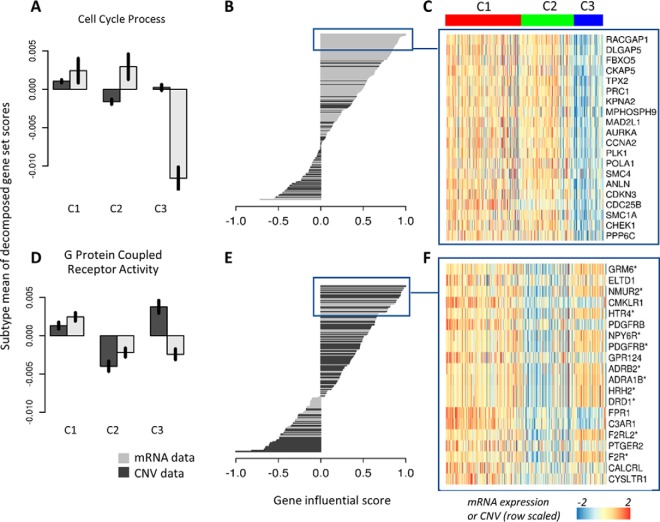
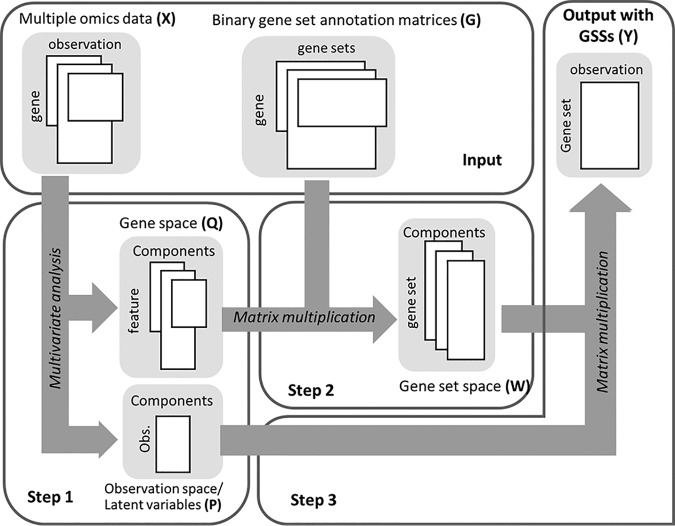
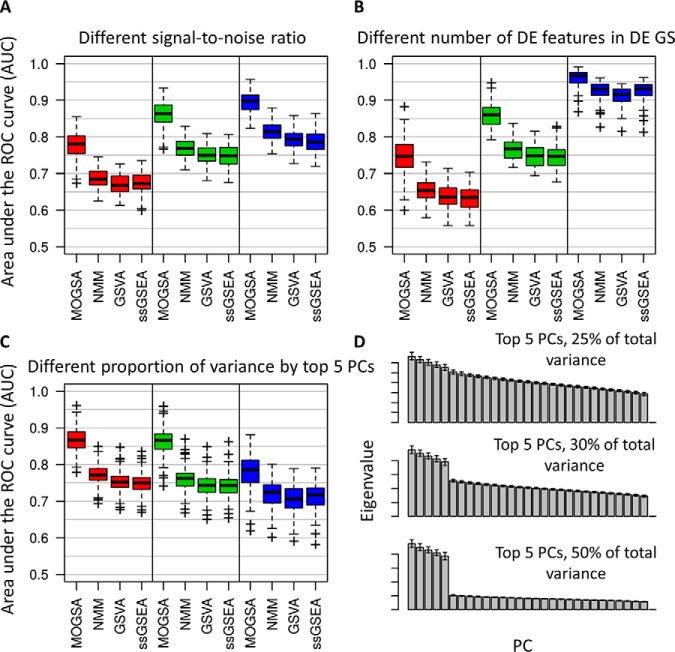
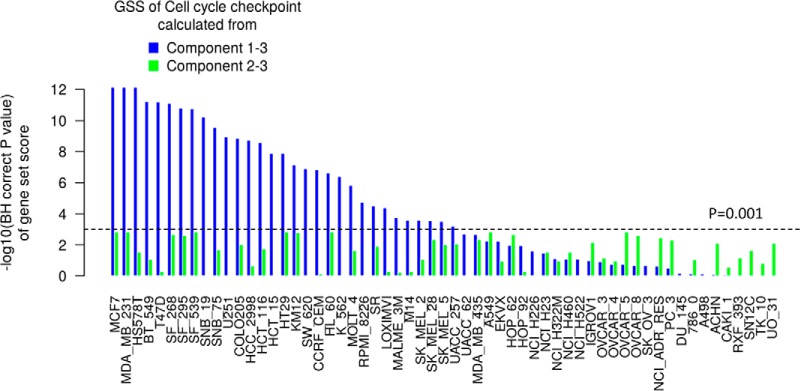
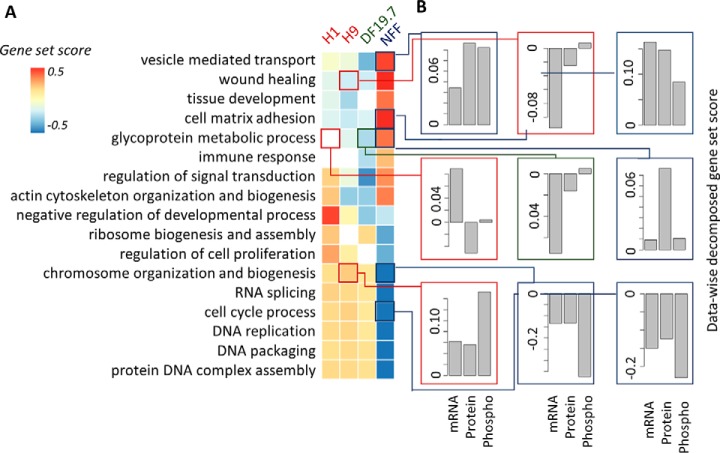
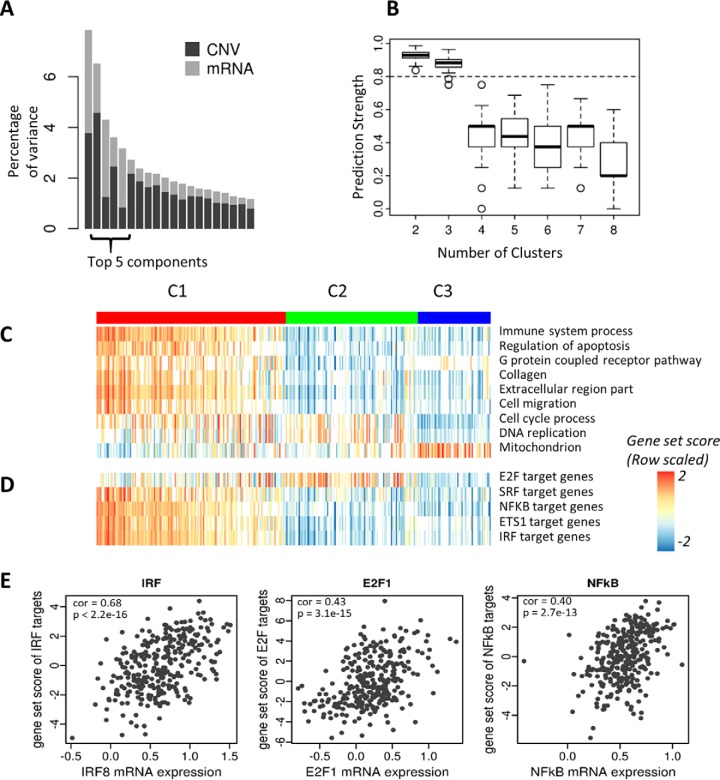
Gene-set analysis (GSA) summarizes individual molecular measurements to more interpretable pathways or gene-sets and has become an indispensable step in the interpretation of large-scale omics data. However, GSA methods are limited to the analysis of single omics data. Here, we introduce a new computation method termed multi-omics gene-set analysis (MOGSA), a multivariate single sample gene-set analysis method that integrates multiple experimental and molecular data types measured over the same set of samples. The method learns a low dimensional representation of most variant correlated features (genes, proteins, etc.) across multiple omics data sets, transforms the features onto the same scale and calculates an integrated gene-set score from the most informative features in each data type. MOGSA does not require filtering data to the intersection of features (gene IDs), therefore, all molecular features, including those that lack annotation may be included in the analysis. Using simulated data, we demonstrate that integrating multiple diverse sources of molecular data increases the power to discover subtle changes in gene-sets and may reduce the impact of unreliable information in any single data type. Using real experimental data, we demonstrate three use-cases of MOGSA. First, we show how to remove a source of noise (technical or biological) in integrative MOGSA of NCI60 transcriptome and proteome data. Second, we apply MOGSA to discover similarities and differences in mRNA, protein and phosphorylation profiles of a small study of stem cell lines and assess the influence of each data type or feature on the total gene-set score. Finally, we apply MOGSA to cluster analysis and show that three molecular subtypes are robustly discovered when copy number variation and mRNA data of 308 bladder cancers from The Cancer Genome Atlas are integrated using MOGSA. MOGSA is available in the Bioconductor R package "mogsa."
基因集分析(GSA)将个体分子测量结果汇总到更具可解释性的途径或基因集中,已成为解释大规模组学数据不可或缺的步骤。然而,GSA 方法仅限于单一组学数据的分析。在这里,我们引入了一种新的计算方法,称为多组学基因集分析(MOGSA),这是一种多元单样本基因集分析方法,可整合在同一组样本上测量的多个实验和分子数据类型。该方法学习了多个组学数据集中大多数与变体相关的特征(基因、蛋白质等)的低维表示形式,将特征转换到同一尺度,并从每个数据类型中最具信息量的特征计算综合基因集得分。MOGSA 不需要将数据过滤到特征(基因 ID)的交集,因此,所有分子特征,包括那些缺乏注释的特征,都可以包含在分析中。使用模拟数据,我们证明了整合多种不同来源的分子数据可以提高发现基因集细微变化的能力,并可能减少任何单一数据类型中不可靠信息的影响。使用真实的实验数据,我们展示了 MOGSA 的三个用例。首先,我们展示了如何在 NCI60 转录组和蛋白质组数据的集成 MOGSA 中去除噪声源(技术或生物学)。其次,我们应用 MOGSA 来发现干细胞系小研究中的 mRNA、蛋白质和磷酸化谱的相似性和差异,并评估每种数据类型或特征对总基因集得分的影响。最后,我们应用 MOGSA 进行聚类分析,并展示了当整合来自癌症基因组图谱的 308 例膀胱癌的拷贝数变异和 mRNA 数据时,三个分子亚型是如何稳健地被发现的。MOGSA 可在 Bioconductor R 包“mogsa”中使用。