Departments of Human Anatomy and Cell Science, University of Manitoba, Winnipeg, Canada.
The Diabetes Research Envisioned and Accomplished in Manitoba (DREAM) Theme, University of Manitoba, Winnipeg, Canada.
Autophagy. 2021 Sep;17(9):2257-2272. doi: 10.1080/15548627.2020.1821548. Epub 2020 Oct 12.
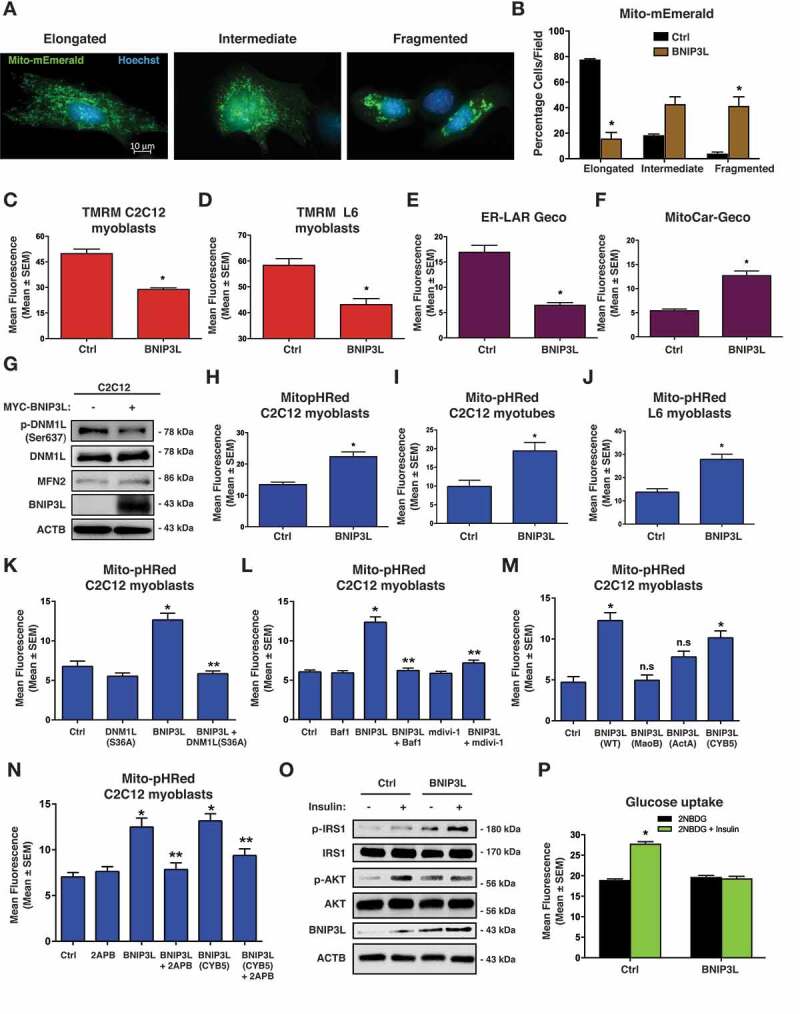
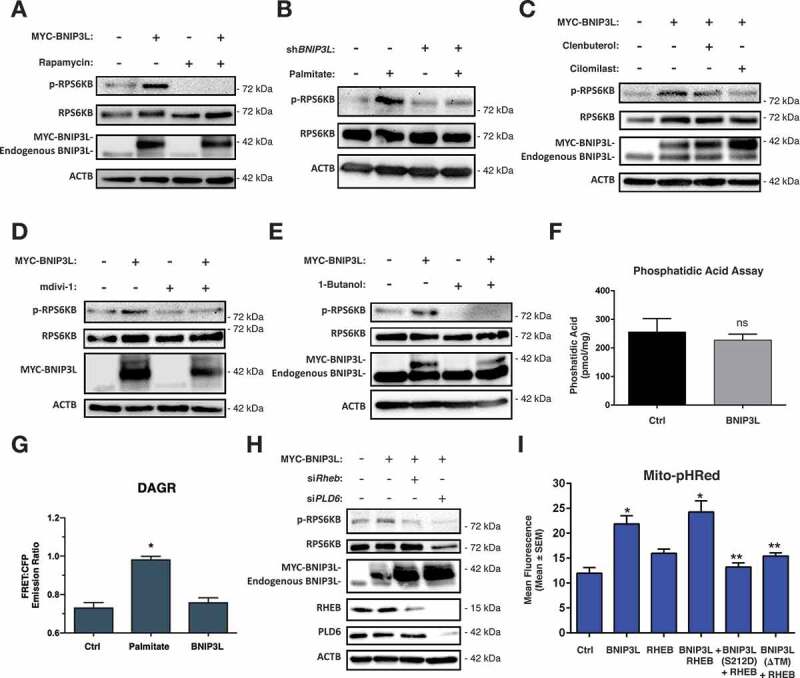
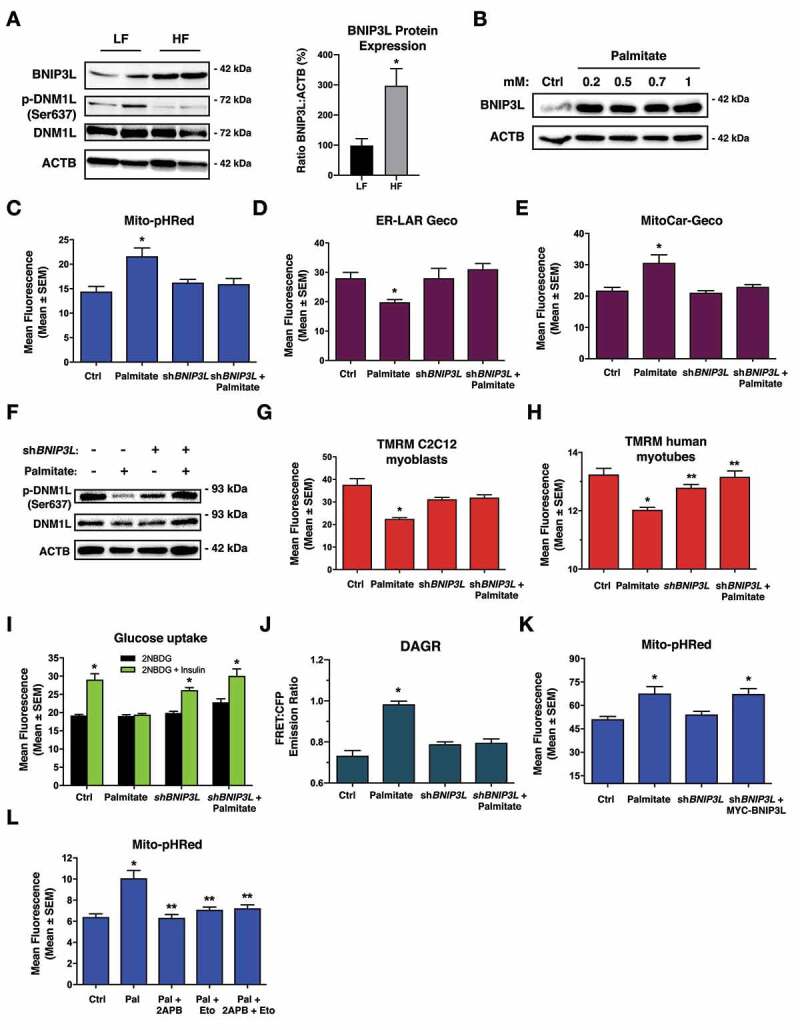
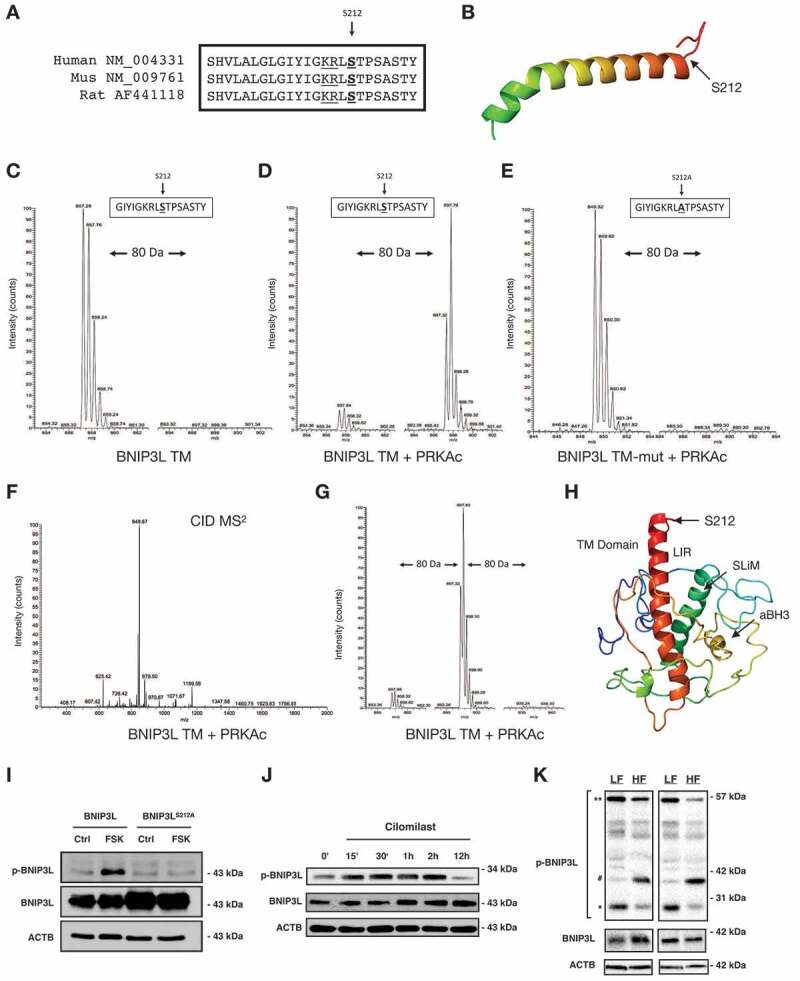
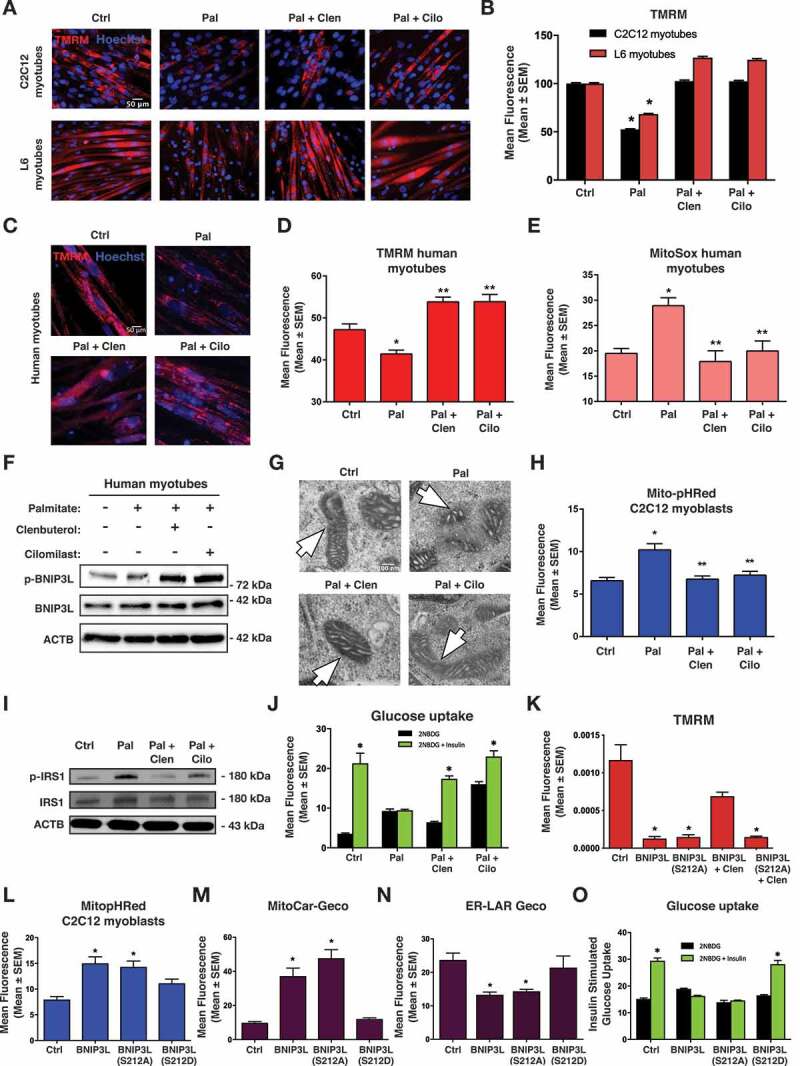
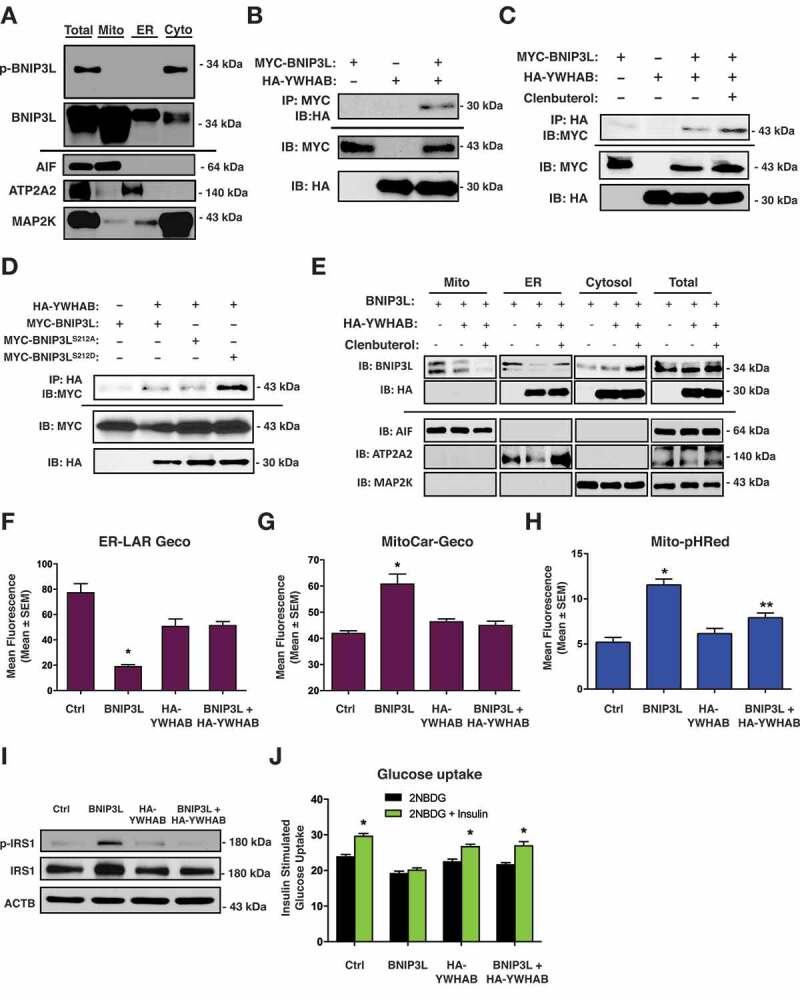
Lipotoxicity is a form of cellular stress caused by the accumulation of lipids resulting in mitochondrial dysfunction and insulin resistance in muscle. Previously, we demonstrated that the mitophagy receptor BNIP3L/Nix is responsive to lipotoxicity and accumulates in response to a high-fat (HF) feeding. To provide a better understanding of this observation, we undertook gene expression array and shot-gun metabolomics studies in soleus muscle from rodents on an HF diet. Interestingly, we observed a modest reduction in several autophagy-related genes. Moreover, we observed alterations in the fatty acyl composition of cardiolipins and phosphatidic acids. Given the reported roles of these phospholipids and BNIP3L in mitochondrial dynamics, we investigated aberrant mitochondrial turnover as a mechanism of impaired myocyte insulin signaling. In a series of gain-of-function and loss-of-function experiments in rodent and human myotubes, we demonstrate that BNIP3L accumulation triggers mitochondrial depolarization, calcium-dependent activation of DNM1L/DRP1, and mitophagy. In addition, BNIP3L can inhibit insulin signaling through activation of MTOR-RPS6KB/p70S6 kinase inhibition of IRS1, which is contingent on phosphatidic acids and RHEB. Finally, we demonstrate that BNIP3L-induced mitophagy and impaired glucose uptake can be reversed by direct phosphorylation of BNIP3L by PRKA/PKA, leading to the translocation of BNIP3L from the mitochondria and sarcoplasmic reticulum to the cytosol. These findings provide insight into the role of BNIP3L, mitochondrial turnover, and impaired myocyte insulin signaling during an overfed state when overall autophagy-related gene expression is reduced. Furthermore, our data suggest a mechanism by which exercise or pharmacological activation of PRKA may overcome myocyte insulin resistance. BCL2: B cell leukemia/lymphoma 2; BNIP3L/Nix: BCL2/adenovirus E1B interacting protein 3-like; DNM1L/DRP1: dynamin 1-like; FUNDC1: FUN14 domain containing 1; IRS1: insulin receptor substrate 1; MAP1LC3A/LC3: microtubule-associated protein 1 light chain 3 alpha; MFN1: mitofusin 1; MFN2: mitofusin 2; MTOR: mechanistic target of rapamycin kinase; OPA1: OPA1 mitochondrial dynamin like GTPase; PDE4i: phosphodiesterase 4 inhibitor; PLD1: phospholipase D1; PLD6: phospholipase D family member 6; PRKA/PKA: protein kinase, AMP-activated; PRKCD/PKCδ: protein kinase C, delta; PRKCQ/PKCθ: protein kinase C, theta; RHEB: Ras homolog enriched in brain; RPS6KB/p70S6K: ribosomal protein S6 kinase; SQSTM1/p62: sequestosome 1; YWHAB/14-3-3β: tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein beta.
脂毒性是一种由脂质积累引起的细胞应激形式,导致肌肉中线粒体功能障碍和胰岛素抵抗。此前,我们证明了自噬受体 BNIP3L/Nix 对脂毒性有反应,并在高脂肪(HF)喂养时积累。为了更好地理解这一观察结果,我们对 HF 饮食的啮齿动物的比目鱼肌进行了基因表达谱和鸟枪法代谢组学研究。有趣的是,我们观察到几种自噬相关基因的适度减少。此外,我们观察到心磷脂和磷酸脂酰基酸的脂肪酸组成发生了改变。鉴于这些磷脂和 BNIP3L 在线粒体动力学中的报告作用,我们研究了异常的线粒体周转作为肌细胞胰岛素信号受损的机制。在一系列啮齿动物和人类肌管的功能获得和功能丧失实验中,我们证明 BNIP3L 的积累会引发线粒体去极化、钙依赖性 DNM1L/DRP1 的激活和线粒体自噬。此外,BNIP3L 可以通过激活 MTOR-RPS6KB/p70S6 激酶抑制 IRS1 来抑制胰岛素信号,这取决于磷酸脂酰基酸和 RHEB。最后,我们证明 BNIP3L 诱导的线粒体自噬和葡萄糖摄取受损可以通过 PRKA/PKA 对 BNIP3L 的直接磷酸化来逆转,导致 BNIP3L 从线粒体和肌浆网转移到细胞质。这些发现为 BNIP3L、线粒体周转和过度喂养状态下肌细胞胰岛素信号受损时的自噬相关基因表达总体减少提供了深入了解。此外,我们的数据表明,PRKA/PKA 的运动或药理学激活可能克服肌细胞胰岛素抵抗的机制。BCL2:B 细胞白血病/淋巴瘤 2;BNIP3L/Nix:BCL2/腺病毒 E1B 相互作用蛋白 3 样;DNM1L/DRP1:dynamin 1 样;FUNDC1:FUN14 结构域包含 1;IRS1:胰岛素受体底物 1;MAP1LC3A/LC3:微管相关蛋白 1 轻链 3 alpha;MFN1:线粒体融合蛋白 1;MFN2:线粒体融合蛋白 2;MTOR:雷帕霉素靶蛋白激酶;OPA1:OPA1 线粒体动力蛋白样 GTPase;PDE4i:磷酸二酯酶 4 抑制剂;PLD1:磷脂酶 D1;PLD6:磷脂酶 D 家族成员 6;PRKA/PKA:蛋白激酶 AMP 激活;PRKCD/PKCδ:蛋白激酶 C,delta;PRKCQ/PKCθ:蛋白激酶 C,theta;RHEB:富含脑的 Ras 同源物;RPS6KB/p70S6K:核糖体蛋白 S6 激酶;SQSTM1/p62:自噬体相关蛋白 1;YWHAB/14-3-3β:酪氨酸 3-单加氧酶/色氨酸 5-单加氧酶激活蛋白 beta。