Kimanius Dari, Zickert Gustav, Nakane Takanori, Adler Jonas, Lunz Sebastian, Schönlieb Carola-Bibiane, Öktem Ozan, Scheres Sjors H W
MRC Laboratory of Molecular Biology, Cambridge, United Kingdom.
Department of Mathematics, Royal Institute of Technology (KTH), Sweden.
IUCrJ. 2021 Jan 1;8(Pt 1):60-75. doi: 10.1107/S2052252520014384.
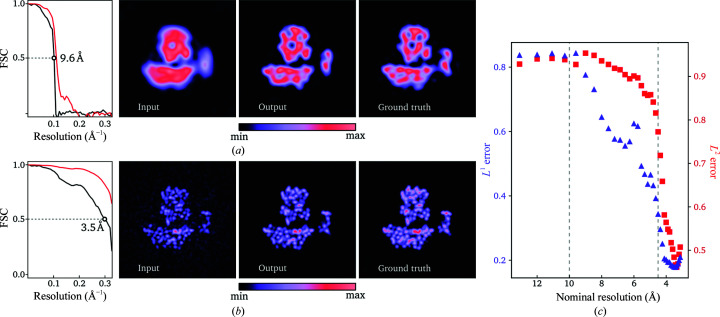
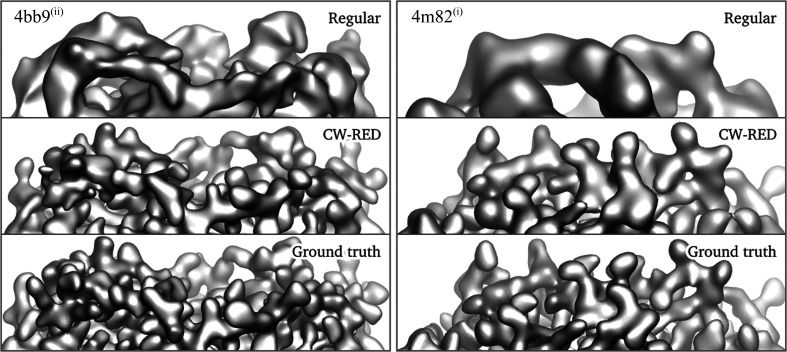
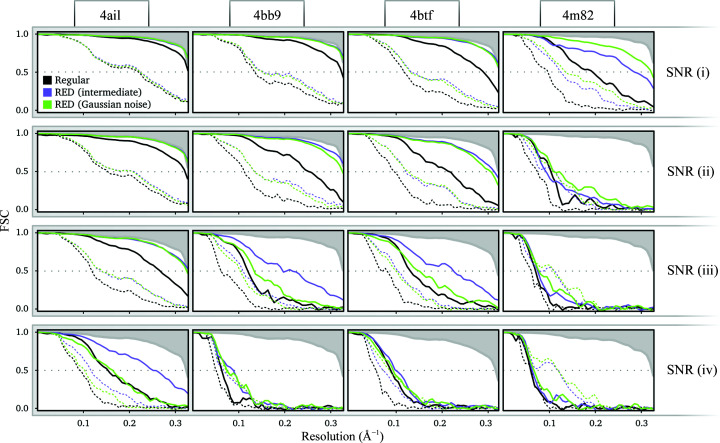
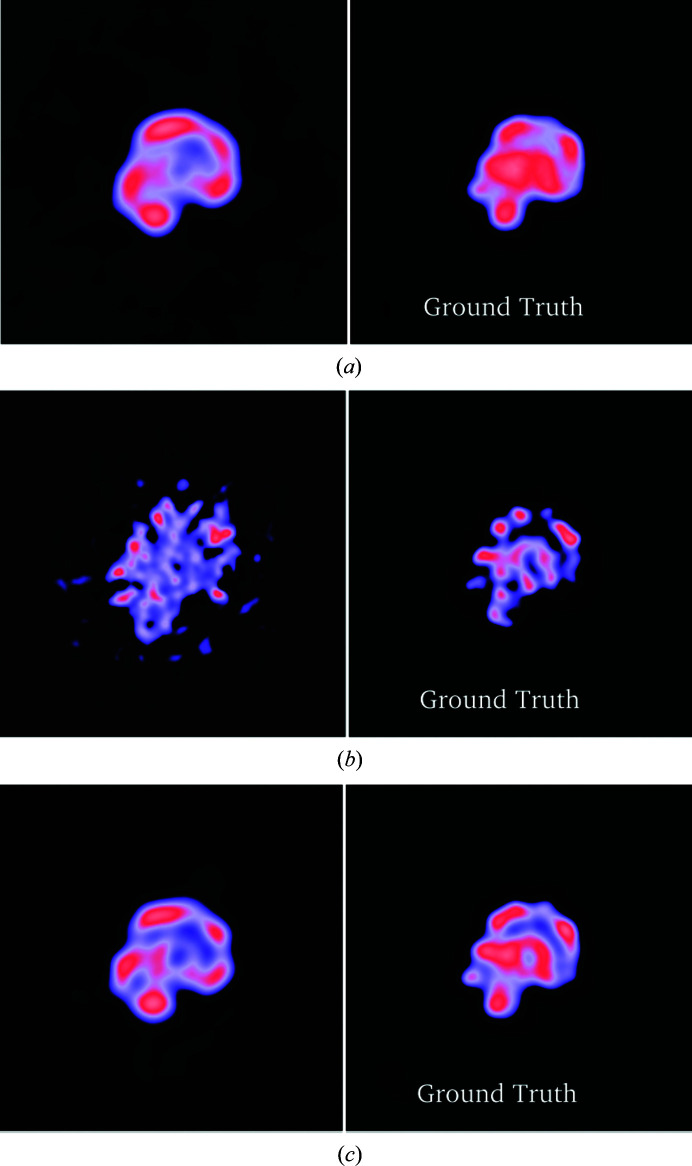
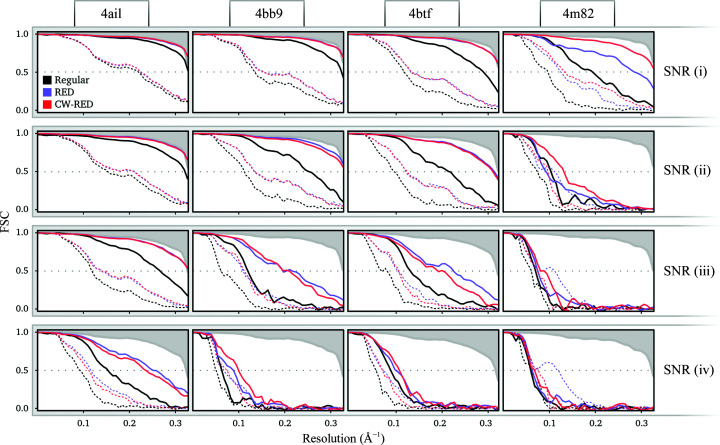
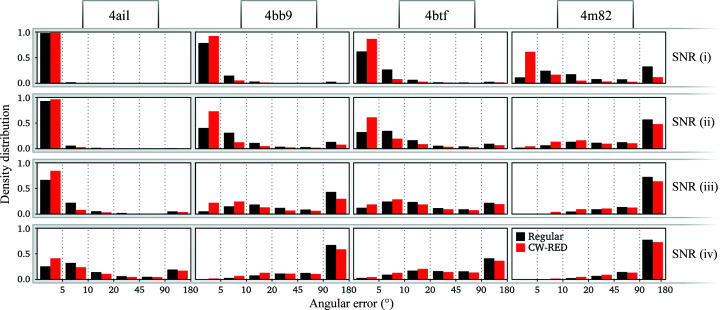
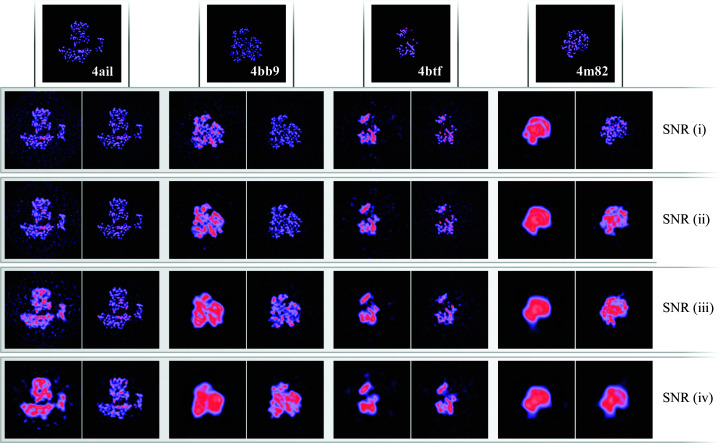
Three-dimensional reconstruction of the electron-scattering potential of biological macromolecules from electron cryo-microscopy (cryo-EM) projection images is an ill-posed problem. The most popular cryo-EM software solutions to date rely on a regularization approach that is based on the prior assumption that the scattering potential varies smoothly over three-dimensional space. Although this approach has been hugely successful in recent years, the amount of prior knowledge that it exploits compares unfavorably with the knowledge about biological structures that has been accumulated over decades of research in structural biology. Here, a regularization framework for cryo-EM structure determination is presented that exploits prior knowledge about biological structures through a convolutional neural network that is trained on known macromolecular structures. This neural network is inserted into the iterative cryo-EM structure-determination process through an approach that is inspired by regularization by denoising. It is shown that the new regularization approach yields better reconstructions than the current state of the art for simulated data, and options to extend this work for application to experimental cryo-EM data are discussed.
从电子冷冻显微镜(cryo-EM)投影图像中对生物大分子的电子散射势进行三维重建是一个不适定问题。迄今为止,最流行的cryo-EM软件解决方案依赖于一种正则化方法,该方法基于散射势在三维空间中平滑变化的先验假设。尽管这种方法近年来取得了巨大成功,但与结构生物学数十年研究积累的有关生物结构的知识相比,它所利用的先验知识量并不占优势。在此,提出了一种用于cryo-EM结构确定的正则化框架,该框架通过在已知大分子结构上训练的卷积神经网络利用有关生物结构的先验知识。通过一种受去噪正则化启发的方法,将该神经网络插入到迭代cryo-EM结构确定过程中。结果表明,对于模拟数据,新的正则化方法比当前的技术水平能产生更好的重建效果,并且还讨论了将这项工作扩展应用于实验cryo-EM数据的方案。