Unidad de Virología, Centro de Educación Médica e Investigaciones Clínicas (CEMIC) Hospital Universitario, Ciudad de Buenos Aires, Argentina.
Departamento de Microbiología, Inmunología y Biotecnología, Cátedra de Virología, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Buenos Aires, Argentina.
PLoS One. 2021 Mar 8;16(3):e0248191. doi: 10.1371/journal.pone.0248191. eCollection 2021.
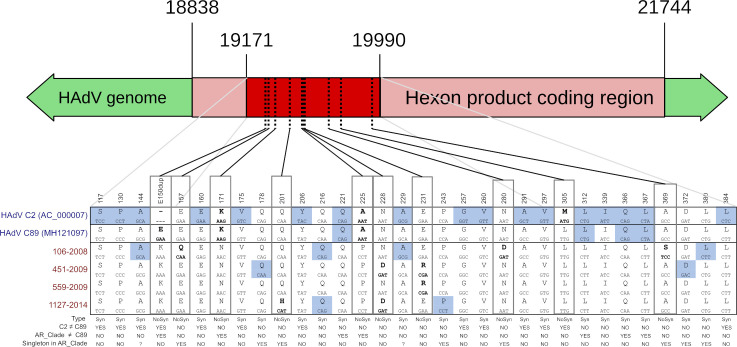
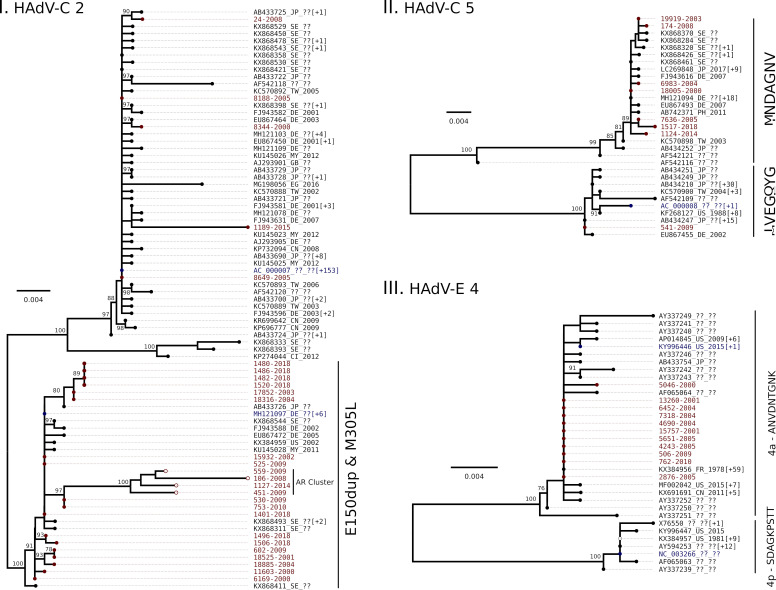
Human adenoviruses (HAdV) are one of the most frequent causes of respiratory infections around the world, causing mild to severe disease. In Argentina, many studies focused on the association of HAdV respiratory infection with severe disease and fatal outcomes leading to the discovery in 1984 of a genomic variant 7h associated with high fatality. Although several molecular studies reported the presence of at least 4 HAdV species (B, C, D and E) in Argentina, few sequences were available in the databases. In this study, sequences from the hexon gene region were obtained from 141 patients as a first approach to assess the genetic diversity of HAdVs circulating in Buenos Aires, Argentina. Phylogenetic analysis of these sequences and others recovered from public databases confirmed the circulation of the four above-mentioned species represented by 11 genotypes, with predominance in species B and C and shifts in their proportion in the studied period (2000 to 2018). The variants detected in Argentina, for most of the genotypes, were similar to those already described in other countries. However, uncommon lineages belonging to genotypes C2, C5 and E4 were detected, which might indicate the circulation of local variants and will deserve further studies of whole-genome sequences.
人腺病毒(HAdV)是世界范围内引起呼吸道感染的最常见原因之一,可导致轻度至重度疾病。在阿根廷,许多研究都集中在 HAdV 呼吸道感染与严重疾病和致命结局的关联上,这导致 1984 年发现了与高死亡率相关的 7h 基因组变异体。尽管有几项分子研究报告称,阿根廷至少存在 4 种 HAdV 种(B、C、D 和 E),但数据库中可用的序列很少。在这项研究中,从 141 名患者中获得了六邻体基因区域的序列,作为评估阿根廷布宜诺斯艾利斯流行的 HAdV 遗传多样性的初步方法。对这些序列和从公共数据库中恢复的其他序列进行的系统发育分析证实,上述四个种(由 11 个基因型代表)在循环,其中 B 和 C 种占优势,在研究期间(2000 年至 2018 年)其比例发生了变化。在阿根廷检测到的变体,对于大多数基因型,与已在其他国家描述的变体相似。然而,检测到属于基因型 C2、C5 和 E4 的不常见谱系,这可能表明当地变体的循环,值得进一步研究全基因组序列。