Regeneron Pharmaceuticals, Inc., Tarrytown, NY, 10591, USA.
Cellular Longevity, Inc., San Francisco, CA, 94103, USA.
Commun Biol. 2021 Oct 22;4(1):1218. doi: 10.1038/s42003-021-02739-1.
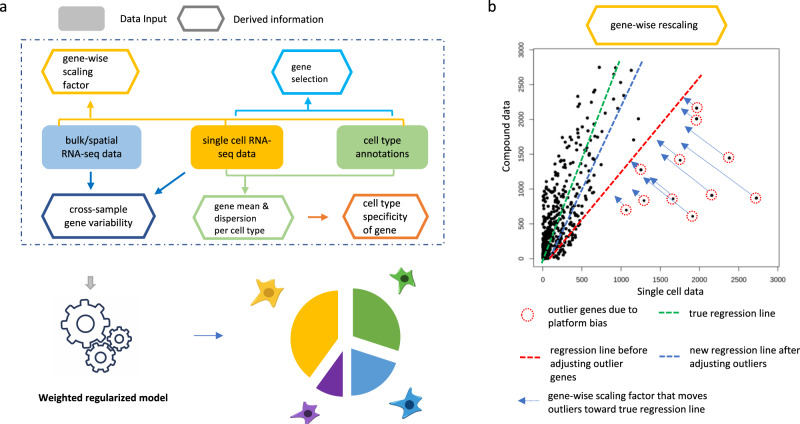
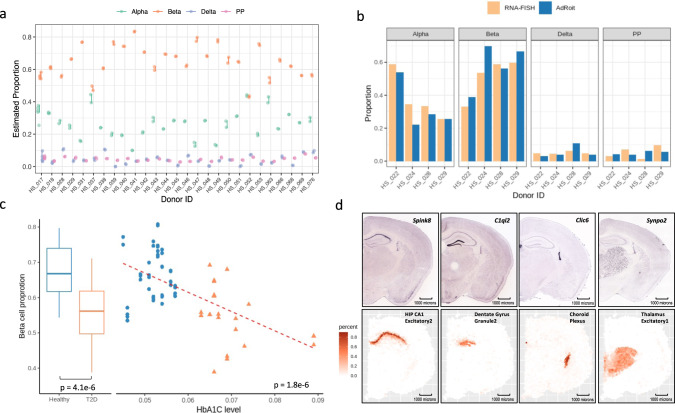
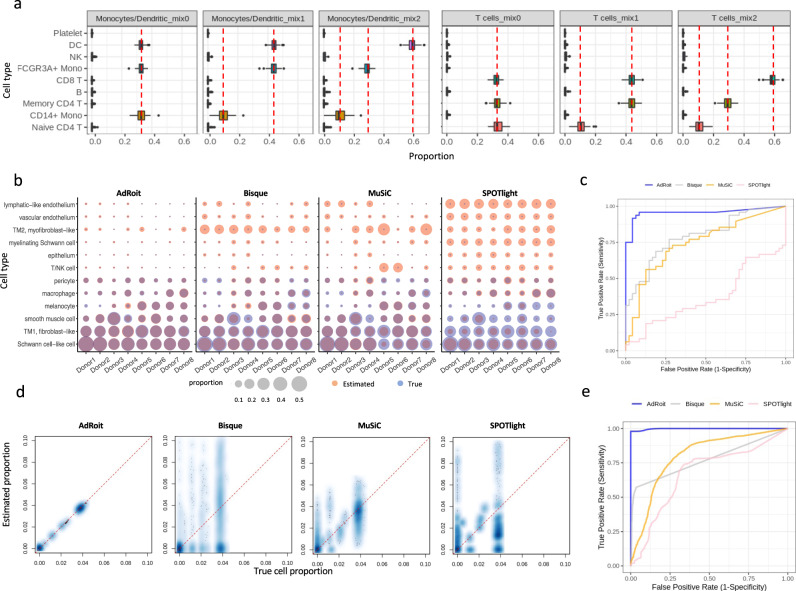
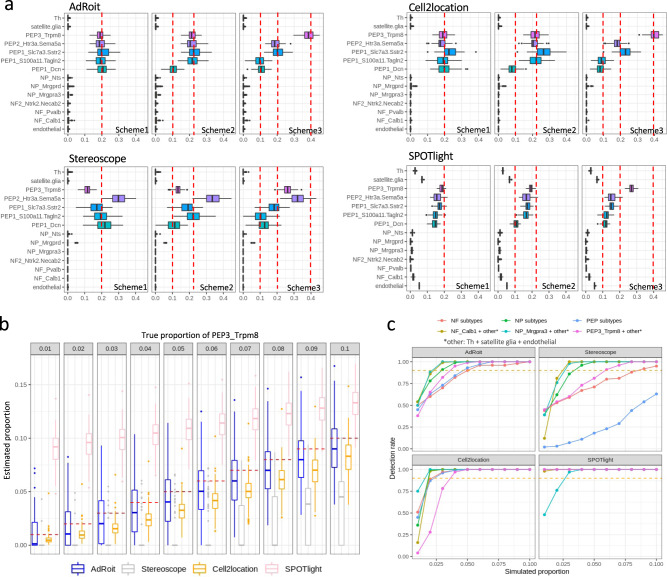
Bulk RNA sequencing provides the opportunity to understand biology at the whole transcriptome level without the prohibitive cost of single cell profiling. Advances in spatial transcriptomics enable to dissect tissue organization and function by genome-wide gene expressions. However, the readout of both technologies is the overall gene expression across potentially many cell types without directly providing the information of cell type constitution. Although several in-silico approaches have been proposed to deconvolute RNA-Seq data composed of multiple cell types, many suffer a deterioration of performance in complex tissues. Here we present AdRoit, an accurate and robust method to infer the cell composition from transcriptome data of mixed cell types. AdRoit uses gene expression profiles obtained from single cell RNA sequencing as a reference. It employs an adaptive learning approach to alleviate the sequencing technique difference between the single cell and the bulk (or spatial) transcriptome data, enhancing cross-platform readout comparability. Our systematic benchmarking and applications, which include deconvoluting complex mixtures that encompass 30 cell types, demonstrate its preferable sensitivity and specificity compared to many existing methods as well as its utilities. In addition, AdRoit is computationally efficient and runs orders of magnitude faster than most methods.
批量 RNA 测序提供了在不进行单细胞分析的高成本的情况下,在整个转录组水平理解生物学的机会。空间转录组学的进步能够通过全基因组基因表达来剖析组织的结构和功能。然而,这两种技术的检测结果都是潜在的许多细胞类型的整体基因表达,而没有直接提供细胞类型组成的信息。尽管已经提出了几种用于解卷积由多种细胞类型组成的 RNA-Seq 数据的计算方法,但许多方法在复杂组织中性能都会恶化。在这里,我们提出了 AdRoit,这是一种从混合细胞类型的转录组数据中推断细胞组成的准确而稳健的方法。AdRoit 使用从单细胞 RNA 测序获得的基因表达谱作为参考。它采用自适应学习方法来减轻单细胞和批量(或空间)转录组数据之间测序技术的差异,增强跨平台读出的可比性。我们的系统基准测试和应用,包括解卷积包含 30 种细胞类型的复杂混合物,证明了它与许多现有方法相比具有更好的敏感性和特异性,以及其实用性。此外,AdRoit 在计算上效率高,运行速度比大多数方法快几个数量级。