Zhang Jiexin, Feng Weijing, Li Minghui, Chen Peier, Ning Xiaodong, Ou Caiwen, Chen Minsheng
Department of Cardiology, Laboratory of Heart Center, Zhujiang Hospital, Southern Medical University, Guangzhou, China.
Guangdong Provincial Key Laboratory of Shock and Microcirculation, Guangzhou, China.
Front Cardiovasc Med. 2021 Nov 8;8:737652. doi: 10.3389/fcvm.2021.737652. eCollection 2021.
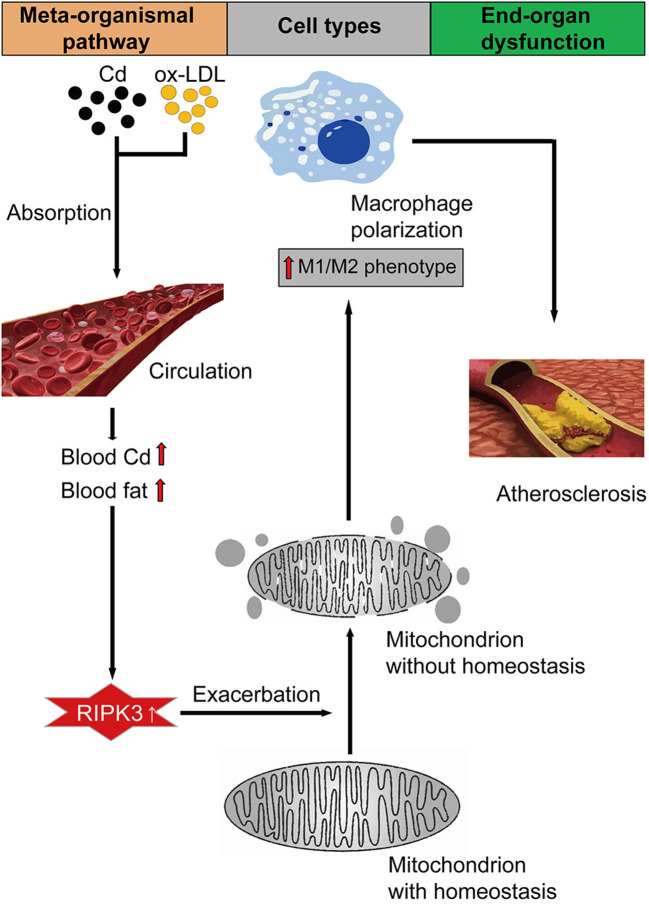
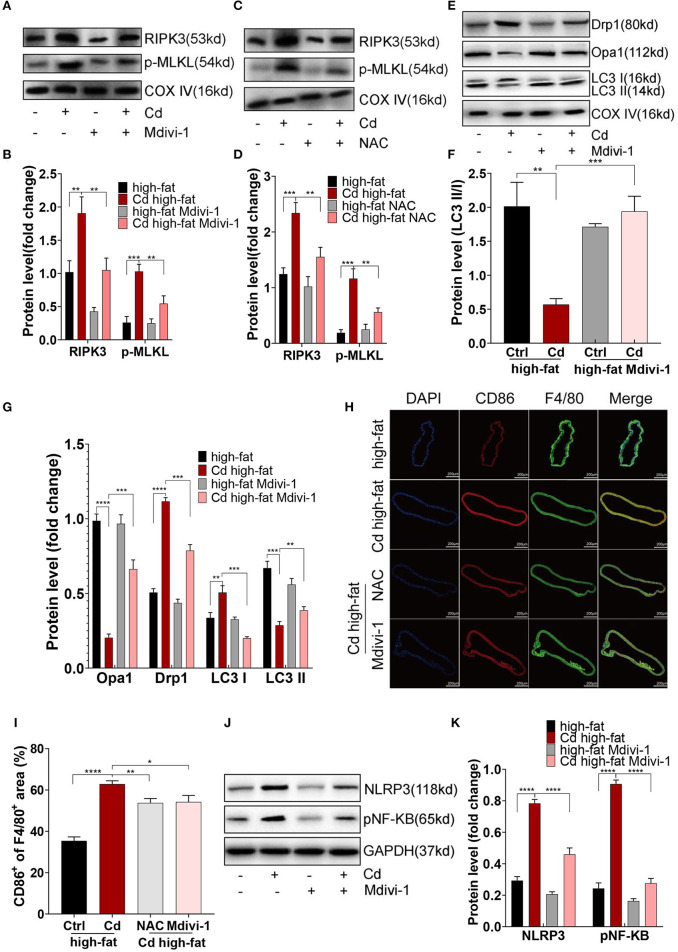
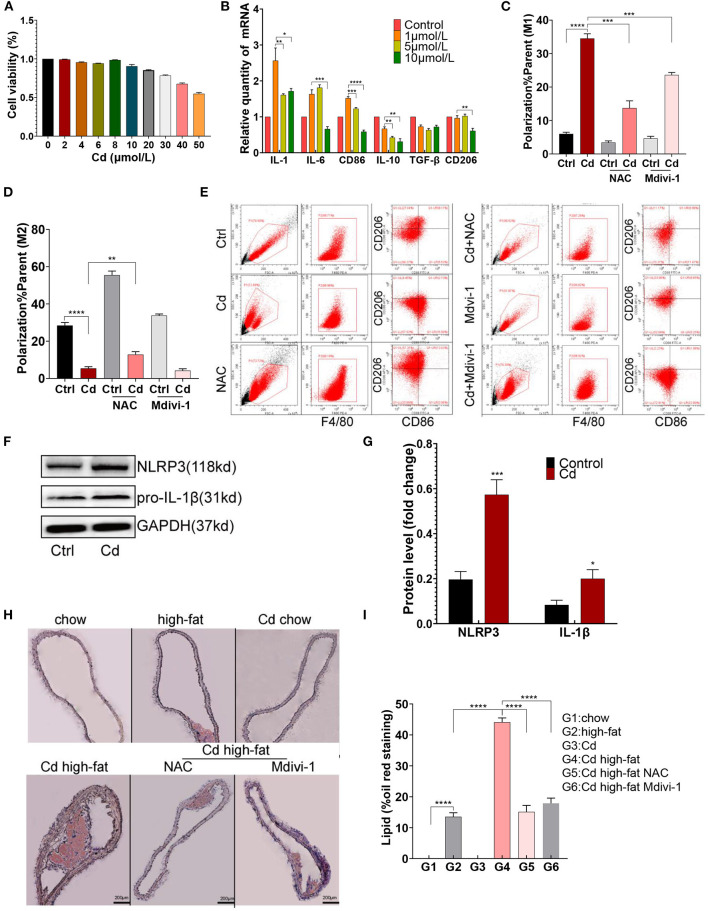
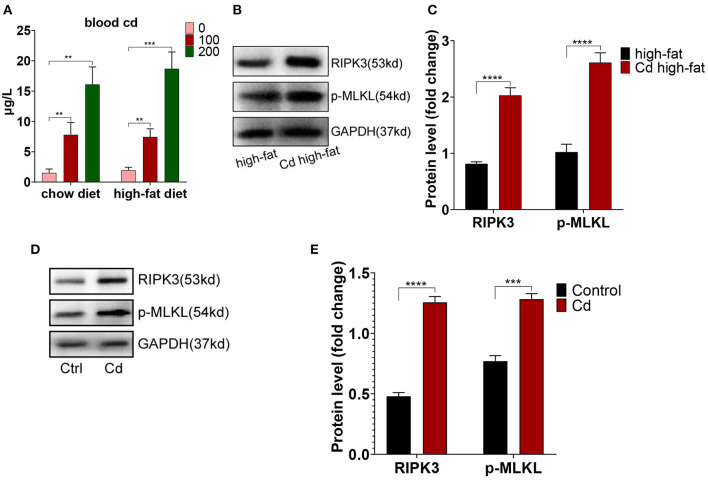
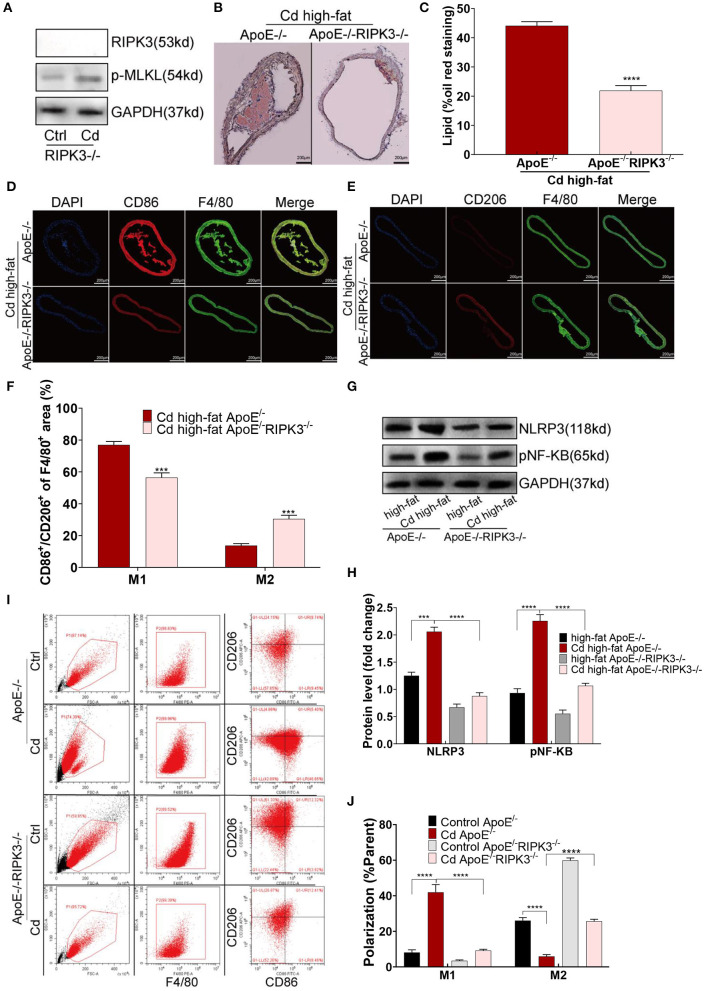
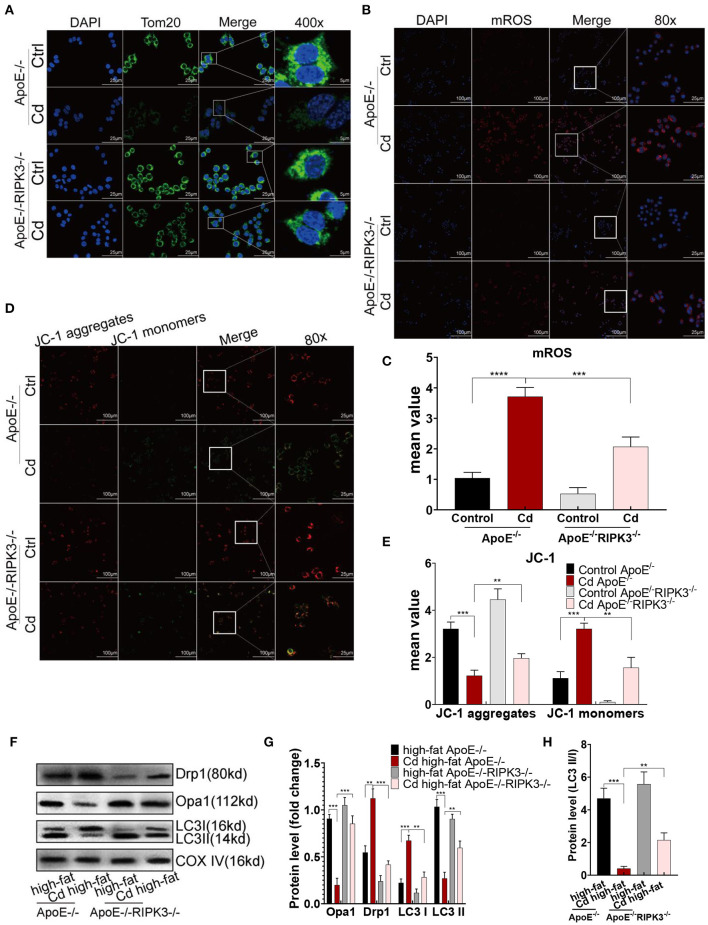
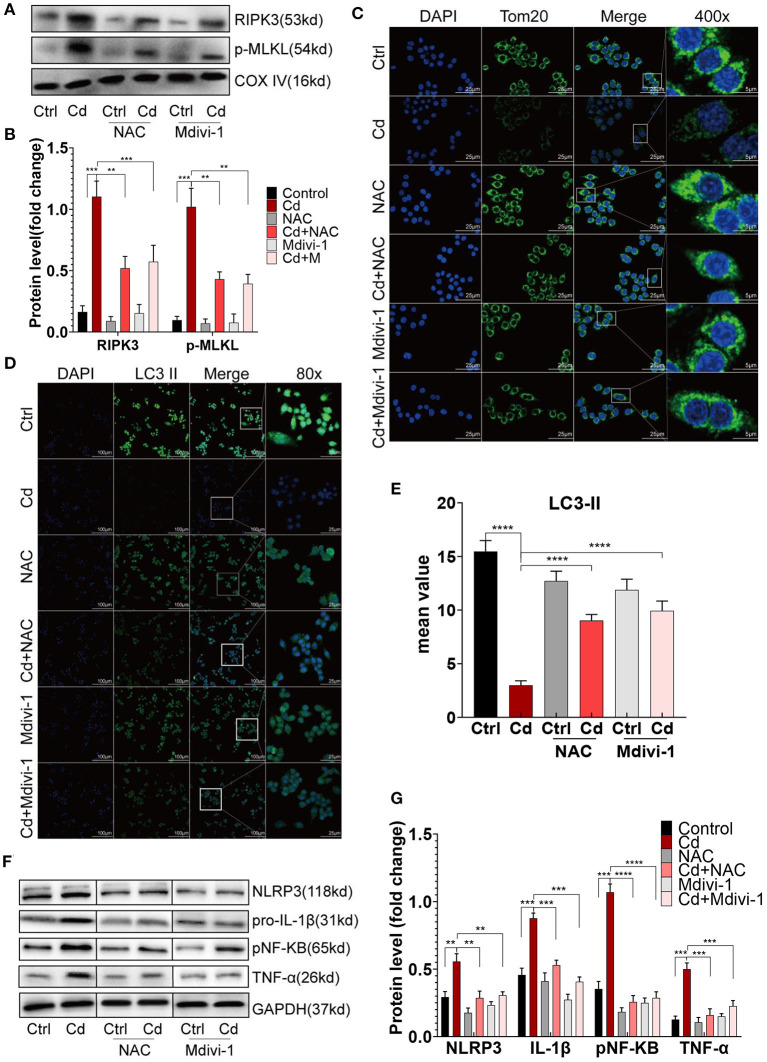
Chronic cadmium (Cd) exposure contributes to the progression of cardiovascular disease (CVD), especially atherosclerosis (AS), but the underlying mechanism is unclear. Since mitochondrial homeostasis is emerging as a core player in the development of CVD, it might serve as a potential mechanism linking Cd exposure and AS. In this study, we aimed to investigate Cd-mediated AS through macrophage polarization and know the mechanisms of Cd-caused mitochondrial homeostasis imbalance. , flow cytometry shows that Cd exposure promotes M1-type polarization of macrophages, manifested as the increasing expressions of nuclear Factor kappa-light-chain-enhancer of activated B (NF-kB) and NLR family pyrin domain containing 3 (NLRP3). Mitochondrial homeostasis tests revealed that decreasing mitochondrial membrane potential and mitophage, increasing the mitochondrial superoxide (mROS), and mitochondrial fission are involved in the Cd-induced macrophage polarization. The upregulated expressions of receptor-interacting protein kinase 3 (RIPK3) and pseudokinase-mixed lineage kinase domain-like protein (p-MLKL) were observed. Knocking out RIPK3, followed by decreasing the expression of p-MLKL, improves the mitochondrial homeostasis imbalance which effectively reverses macrophage polarization. , the oil red O staining showed that Cd with higher blood significantly aggravates AS. Besides, M1-type polarization of macrophages and mitochondrial homeostasis imbalance were observed in the aortic roots of the mice through immunofluorescence and western blot. Knocking out RIPK3 restored the changes above. Finally, the administered N-acetyl cysteine (NAC) or mitochondrial division inhibitor-1 (Mdivi-1), which decreased the mROS or mitochondrial fission, inhibited the expressions of RIPK3 and p-MLKL, attenuating AS and macrophage M1-type polarization in the Cd-treated group. Consequently, the Cd exposure activated the RIPK3 pathway and impaired the mitochondrial homeostasis, resulting in pro-inflammatory macrophage polarization and subsequent AS. Knocking out RIPK3 provided a potential therapeutic target for Cd-caused macrophage polarization and subsequent AS.
长期镉(Cd)暴露会促进心血管疾病(CVD)的发展,尤其是动脉粥样硬化(AS),但其潜在机制尚不清楚。由于线粒体稳态在CVD发展过程中逐渐成为关键因素,它可能是连接Cd暴露与AS的潜在机制。在本研究中,我们旨在通过巨噬细胞极化研究Cd介导的AS,并了解Cd导致线粒体稳态失衡的机制。流式细胞术显示,Cd暴露促进巨噬细胞向M1型极化,表现为活化B细胞核因子κB(NF-κB)和含NLR家族pyrin结构域3(NLRP3)的表达增加。线粒体稳态测试表明,线粒体膜电位降低、线粒体自噬、线粒体超氧化物(mROS)增加以及线粒体分裂参与了Cd诱导的巨噬细胞极化。观察到受体相互作用蛋白激酶3(RIPK3)和假激酶混合谱系激酶结构域样蛋白(p-MLKL)的表达上调。敲除RIPK3,随后降低p-MLKL的表达,改善了线粒体稳态失衡,有效逆转了巨噬细胞极化。油红O染色显示,血液中Cd含量较高会显著加重AS。此外,通过免疫荧光和蛋白质印迹在小鼠主动脉根部观察到巨噬细胞的M1型极化和线粒体稳态失衡。敲除RIPK3可恢复上述变化。最后,给予N-乙酰半胱氨酸(NAC)或线粒体分裂抑制剂-1(Mdivi-1),可降低mROS或线粒体分裂,抑制RIPK3和p-MLKL的表达,减轻Cd处理组的AS和巨噬细胞M1型极化。因此,Cd暴露激活了RIPK3通路并损害了线粒体稳态,导致促炎性巨噬细胞极化和随后的AS。敲除RIPK3为Cd引起的巨噬细胞极化和随后的AS提供了一个潜在的治疗靶点。