Department of Biophysics, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, 14115-154, Iran.
Department of Nanobiotechnology, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, 14115-154, Iran.
Sci Rep. 2022 Feb 11;12(1):2371. doi: 10.1038/s41598-022-06380-8.
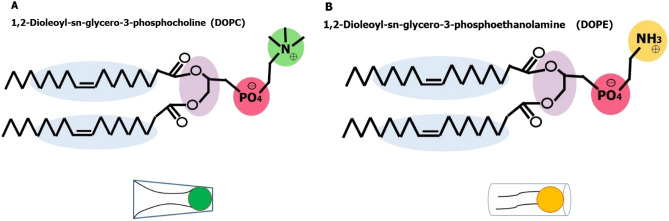
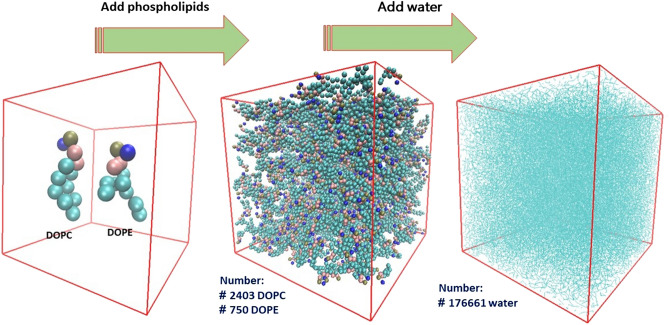
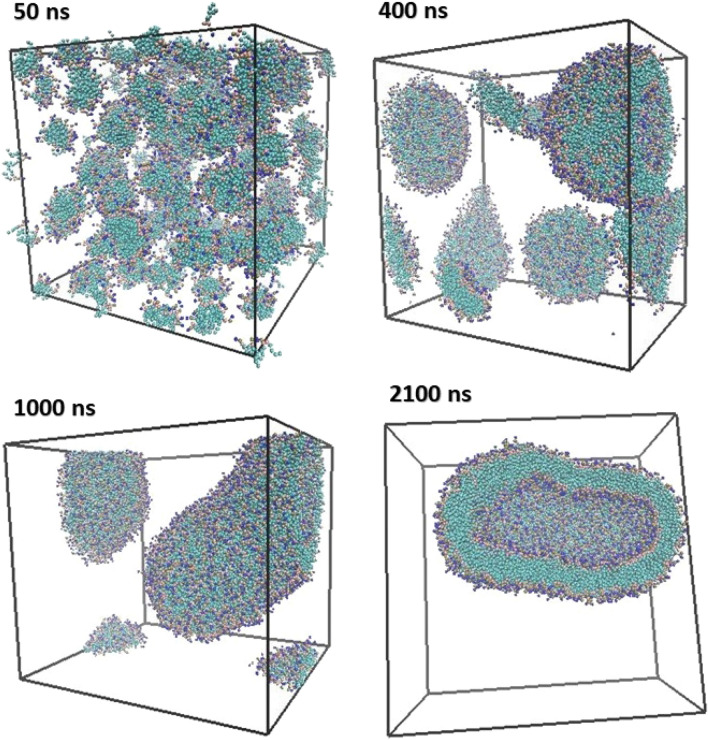
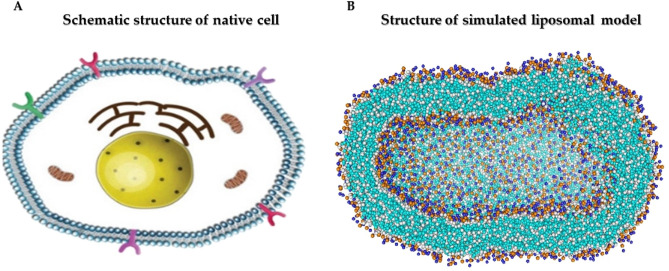
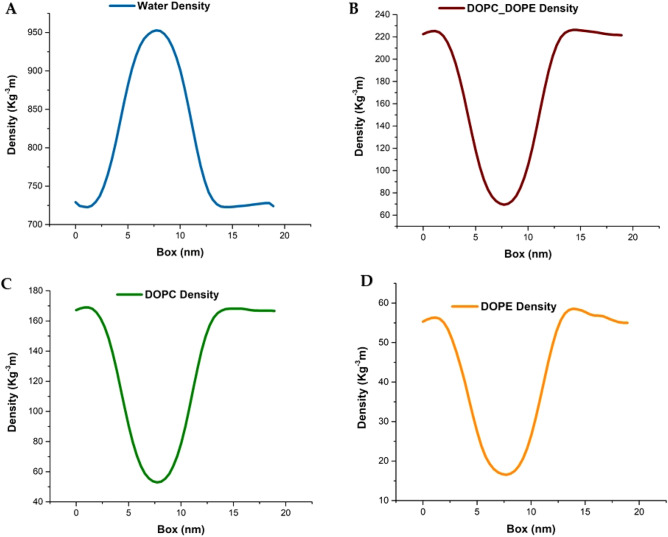
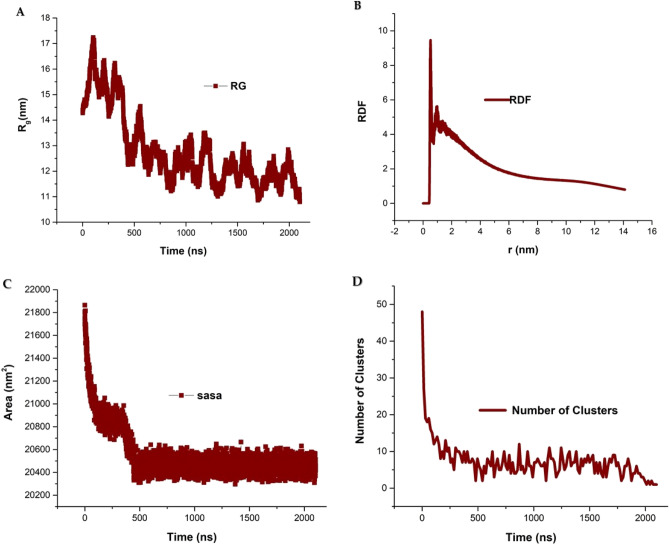
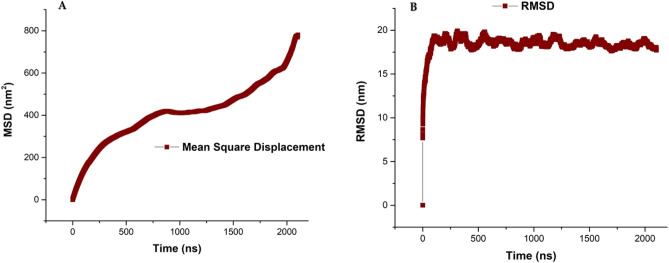
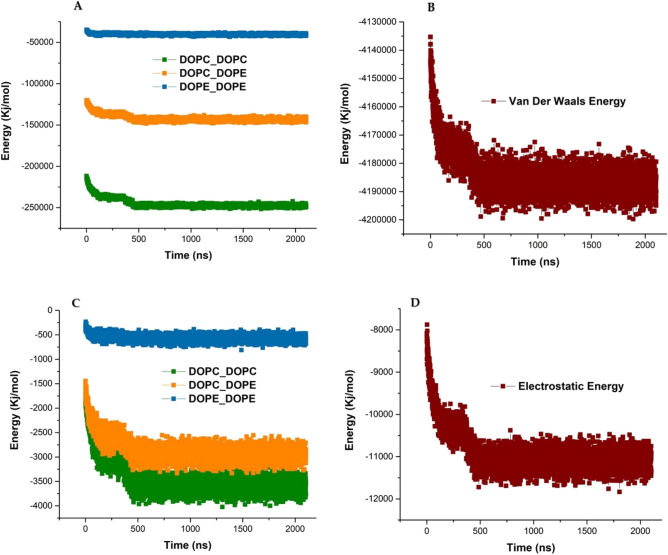
The simulated liposome models provide events in molecular biological science and cellular biology. These models may help to understand the cell membrane mechanisms, biological cell interactions, and drug delivery systems. In addition, the liposomes model may resolve specific issues such as membrane transports, ion channels, drug penetration in the membrane, vesicle formation, membrane fusion, and membrane protein function mechanism. One of the approaches to investigate the lipid membranes and the mechanism of their formation is by molecular dynamics (MD) simulations. In this study, we used the coarse-grained MD simulation approach and designed a liposome model system. To simulate the liposome model, we used phospholipids that are present in the structure of natural cell membranes (1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)). Simulation conditions such as temperature, ions, water, lipid concentration were performed based on experimental conditions. Our results showed a liposome model (ellipse vesicle structure) during the 2100 ns was formed. Moreover, the analysis confirmed that the stretched and ellipse structure is the best structure that could be formed. The eukaryotic and even the bacterial cells have elliptical and flexible structures. Usually, an elliptical structure is more stable than other assembled structures. The results indicated the assembly of the lipids is directed through short-range interactions (electrostatic interactions and, van der Waals interactions). Total energy (Van der Waals and electrostatic interaction energy) confirmed the designed elliptical liposome structure has suitable stability at the end of the simulation process. Our findings confirmed that phospholipids DOPC and DOPE have a good tendency to form bilayer membranes (liposomal structure) based on their geometric shapes and chemical-physical properties. Finally, we expected the simulated liposomal structure as a simple model to be useful in understanding the function and structure of biological cell membranes. Furthermore, it is useful to design optimal, suitable, and biocompatible liposomes as potential drug carriers.
模拟脂质体模型提供了分子生物学科学和细胞生物学领域的事件。这些模型可以帮助理解细胞膜机制、生物细胞相互作用和药物输送系统。此外,脂质体模型还可以解决特定问题,如膜转运、离子通道、药物在膜中的渗透、囊泡形成、膜融合和膜蛋白功能机制。研究脂质膜及其形成机制的一种方法是通过分子动力学 (MD) 模拟。在这项研究中,我们使用了粗粒 MD 模拟方法,并设计了一个脂质体模型系统。为了模拟脂质体模型,我们使用了存在于天然细胞膜结构中的磷脂(1,2-二油酰基-sn-甘油-3-磷酸胆碱 (DOPC) 和 1,2-二油酰基-sn-甘油-3-磷酸乙醇胺 (DOPE))。模拟条件,如温度、离子、水、脂质浓度,是根据实验条件进行的。我们的结果显示,在 2100ns 期间形成了一个脂质体模型(椭圆囊泡结构)。此外,分析证实拉伸和椭圆结构是能够形成的最佳结构。真核生物甚至细菌细胞都具有椭圆形和灵活的结构。通常,椭圆形结构比其他组装结构更稳定。结果表明,脂质的组装是通过短程相互作用(静电相互作用和范德华相互作用)来导向的。总能量(范德华和静电相互作用能)证实,在模拟过程结束时,设计的椭圆脂质体结构具有合适的稳定性。我们的研究结果证实,DOPC 和 DOPE 磷脂具有良好的倾向,根据它们的几何形状和物理化学性质形成双层膜(脂质体结构)。最后,我们期望模拟的脂质体结构作为一个简单的模型,有助于理解生物细胞膜的功能和结构。此外,它还可以用于设计最佳、合适和生物相容的脂质体作为潜在的药物载体。