Department of Computer Science and Applied Mathematics, and Department of Immunology and Reproductive Biology, Weizmann Institute of Science, Rehovot, Israel.
Genome Biol. 2022 Apr 19;23(1):100. doi: 10.1186/s13059-022-02667-1.
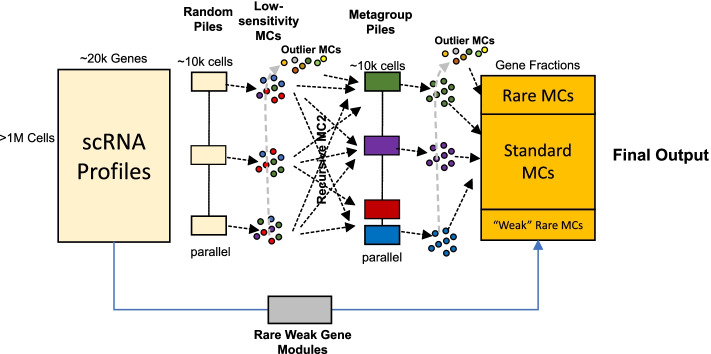
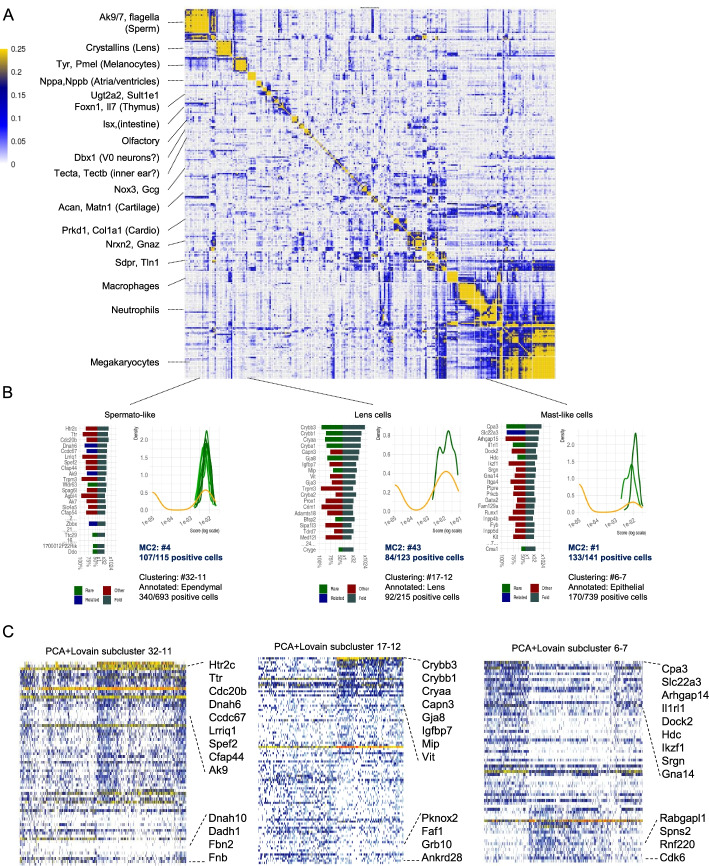
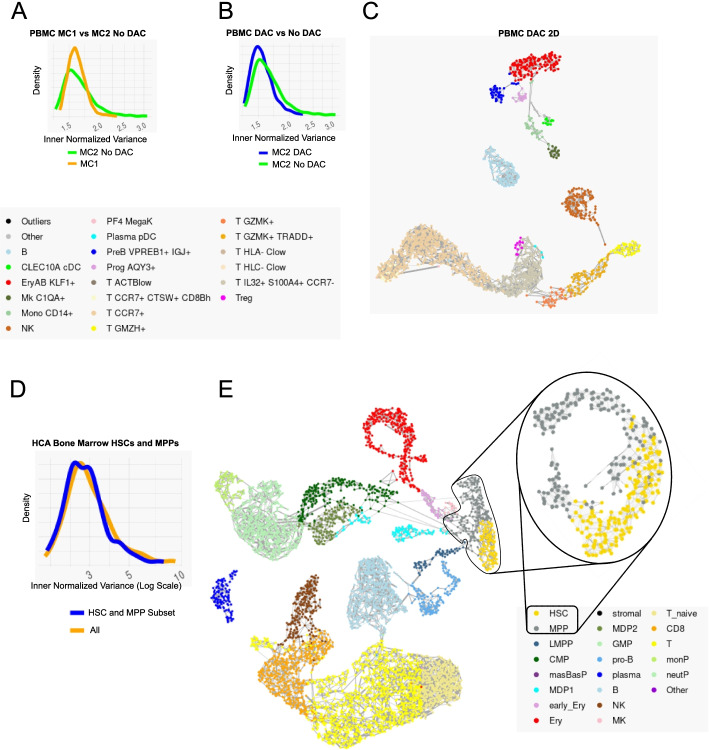
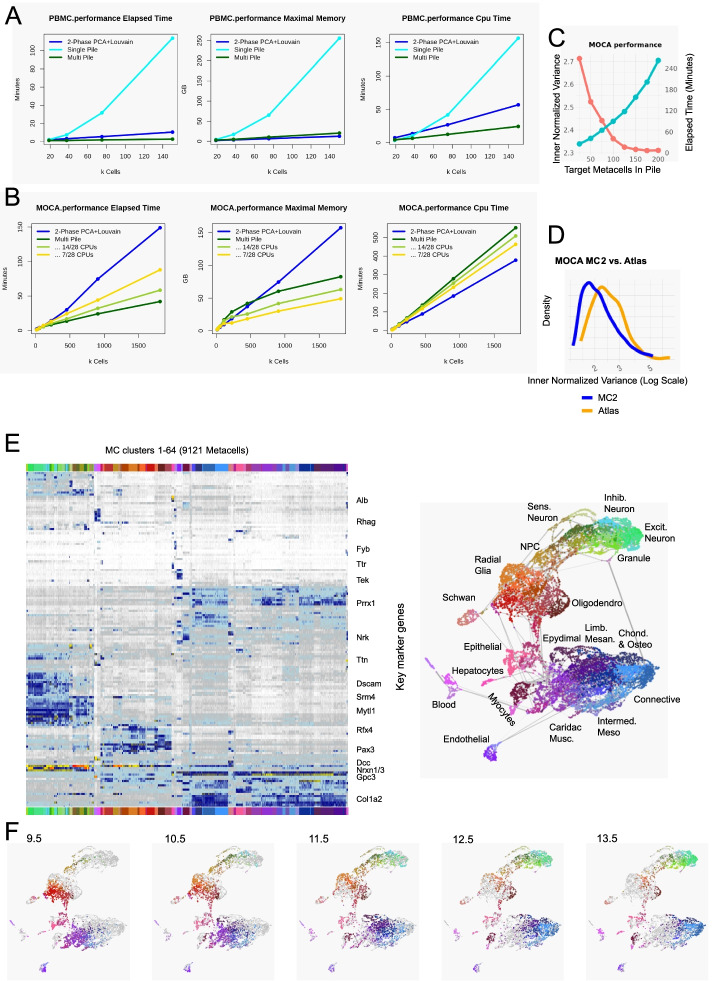
Scaling scRNA-seq to profile millions of cells is crucial for constructing high-resolution maps of transcriptional manifolds. Current analysis strategies, in particular dimensionality reduction and two-phase clustering, offer only limited scaling and sensitivity to define such manifolds. We introduce Metacell-2, a recursive divide-and-conquer algorithm allowing efficient decomposition of scRNA-seq datasets of any size into small and cohesive groups of cells called metacells. Metacell-2 improves outlier cell detection and rare cell type identification, as shown with human bone marrow cell atlas and mouse embryonic data. Metacell-2 is implemented over the scanpy framework for easy integration in any analysis pipeline.
对 scRNA-seq 进行规模化处理以对数百万个细胞进行分析,对于构建转录流形的高分辨率图谱至关重要。当前的分析策略,特别是降维和两阶段聚类,仅提供有限的扩展性和灵敏度来定义此类流形。我们引入了 Metacell-2,这是一种递归的分治算法,可以将任何大小的 scRNA-seq 数据集有效地分解成称为 metacells 的小而有凝聚力的细胞群。Metacell-2 提高了异常细胞检测和稀有细胞类型识别的效果,在人类骨髓细胞图谱和小鼠胚胎数据中得到了验证。Metacell-2 是在 scanpy 框架上实现的,可轻松集成到任何分析管道中。