Molecular Biosciences, Northwestern University, Evanston, IL 60208, USA.
Department of Biological Chemistry, University of California-Los Angeles, Los Angeles, CA 90095, USA.
G3 (Bethesda). 2022 Jul 6;12(7). doi: 10.1093/g3journal/jkac114.
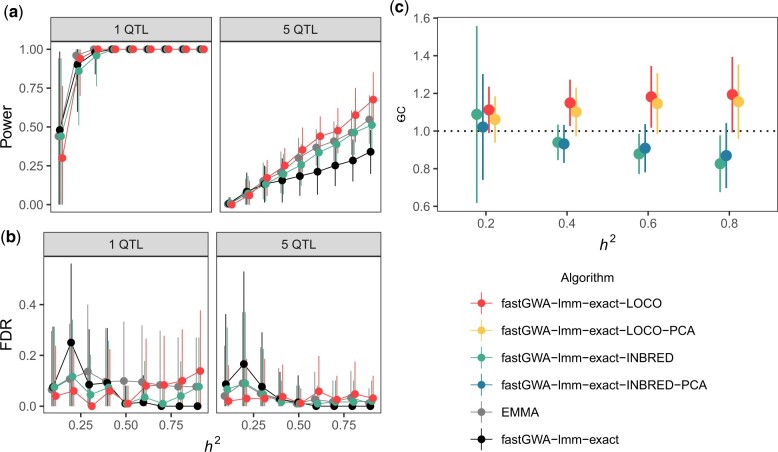
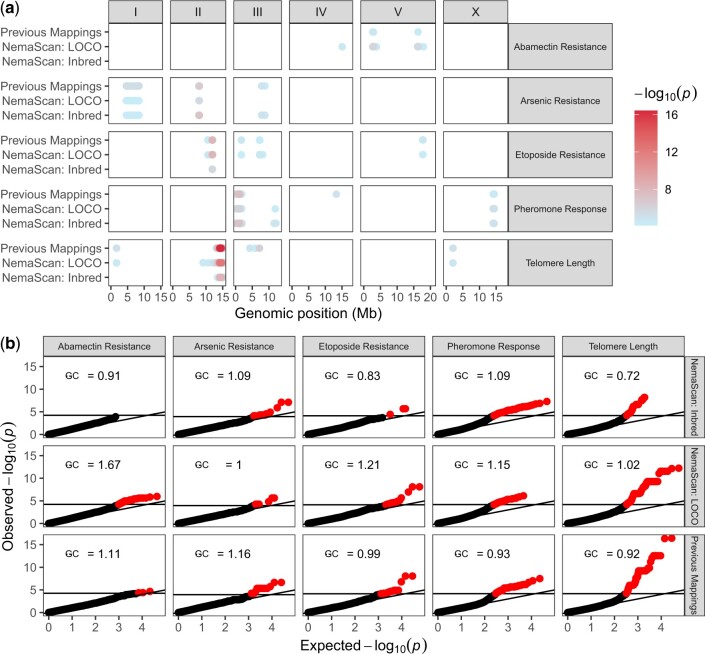
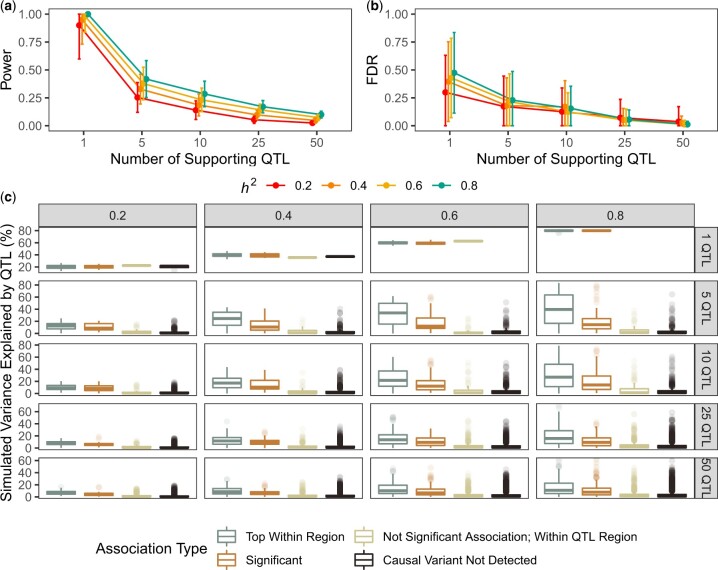
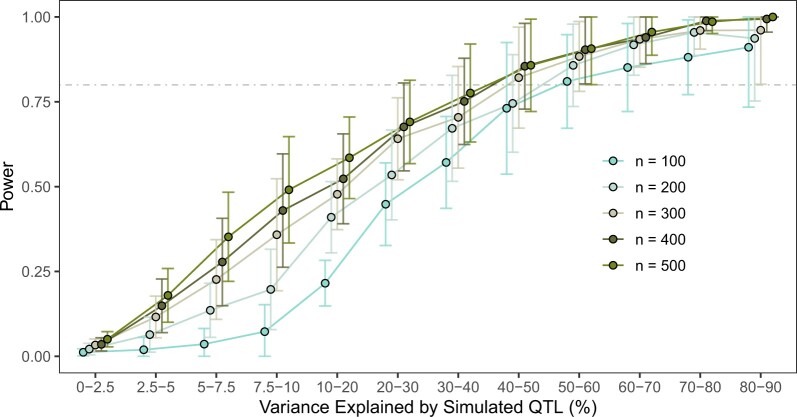
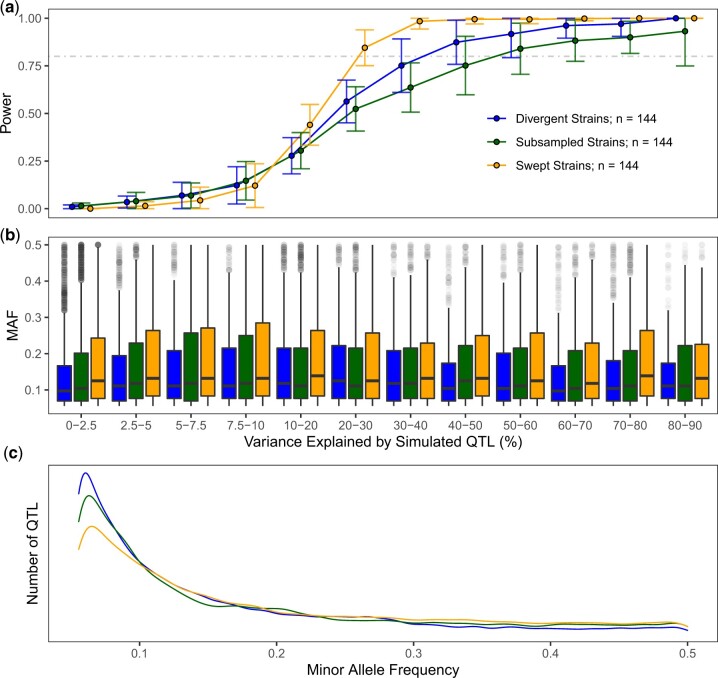
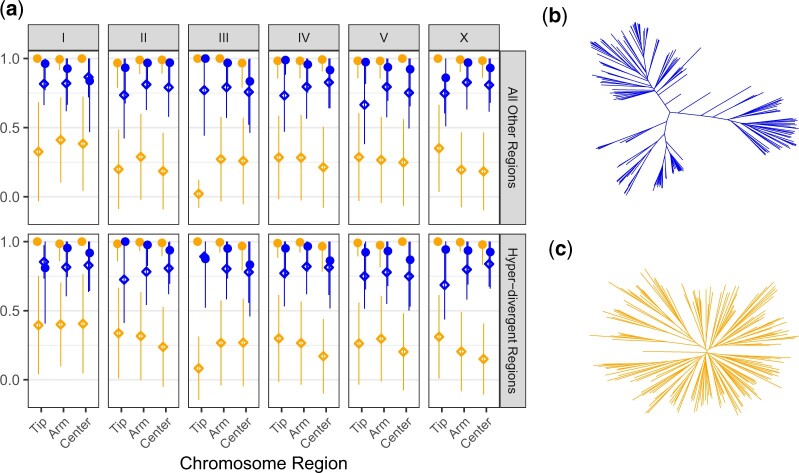
Quantitative genetics in Caenorhabditis elegans seeks to identify naturally segregating genetic variants that underlie complex traits. Genome-wide association studies scan the genome for individual genetic variants that are significantly correlated with phenotypic variation in a population, or quantitative trait loci. Genome-wide association studies are a popular choice for quantitative genetic analyses because the quantitative trait loci that are discovered segregate in natural populations. Despite numerous successful mapping experiments, the empirical performance of genome-wide association study has not, to date, been formally evaluated in C. elegans. We developed an open-source genome-wide association study pipeline called NemaScan and used a simulation-based approach to provide benchmarks of mapping performance in collections of wild C. elegans strains. Simulated trait heritability and complexity determined the spectrum of quantitative trait loci detected by genome-wide association studies. Power to detect smaller-effect quantitative trait loci increased with the number of strains sampled from the C. elegans Natural Diversity Resource. Population structure was a major driver of variation in mapping performance, with populations shaped by recent selection exhibiting significantly lower false discovery rates than populations composed of more divergent strains. We also recapitulated previous genome-wide association studies of experimentally validated quantitative trait variants. Our simulation-based evaluation of performance provides the community with critical context to pursue quantitative genetic studies using the C. elegans Natural Diversity Resource to elucidate the genetic basis of complex traits in C. elegans natural populations.
秀丽隐杆线虫的数量遗传学旨在识别自然分离的遗传变异,这些变异是复杂性状的基础。全基因组关联研究扫描基因组中与群体表型变异显著相关的个体遗传变异,或数量性状位点。全基因组关联研究是定量遗传分析的热门选择,因为发现的数量性状位点在自然种群中分离。尽管有许多成功的映射实验,但到目前为止,全基因组关联研究在秀丽隐杆线虫中的经验表现尚未得到正式评估。我们开发了一个名为 NemaScan 的开源全基因组关联研究管道,并使用基于模拟的方法为野生秀丽隐杆线虫菌株集合中的映射性能提供基准。模拟性状遗传力和复杂性决定了全基因组关联研究检测到的数量性状位点的范围。从秀丽隐杆线虫自然多样性资源中采样的菌株数量越多,检测到较小效应数量性状位点的能力就越强。种群结构是映射性能变化的主要驱动因素,由近期选择形成的种群比由更多不同菌株组成的种群具有显著更低的假发现率。我们还重现了以前对经过实验验证的定量性状变异的全基因组关联研究。我们基于模拟的性能评估为社区提供了重要的背景,以使用秀丽隐杆线虫自然多样性资源进行定量遗传研究,阐明秀丽隐杆线虫自然种群中复杂性状的遗传基础。