Huo Haoyan, Bartel Christopher J, He Tanjin, Trewartha Amalie, Dunn Alexander, Ouyang Bin, Jain Anubhav, Ceder Gerbrand
Department of Materials Science and Engineering, University of California, Berkeley, 210 Hearst Memorial Mining Building, Berkeley, California 94720, United States.
Materials Sciences Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, United States.
Chem Mater. 2022 Aug 23;34(16):7323-7336. doi: 10.1021/acs.chemmater.2c01293. Epub 2022 Aug 5.
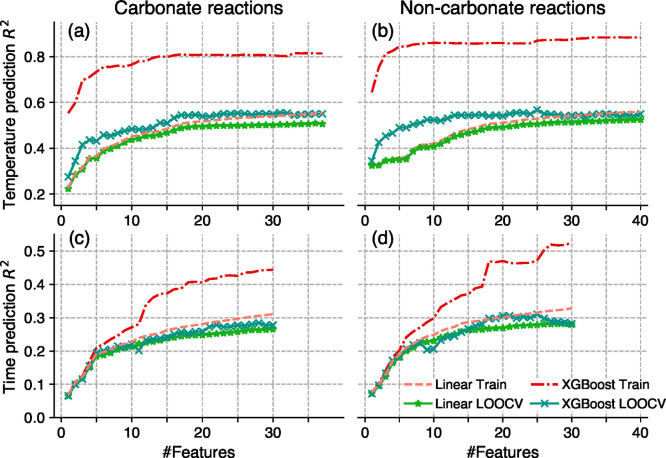
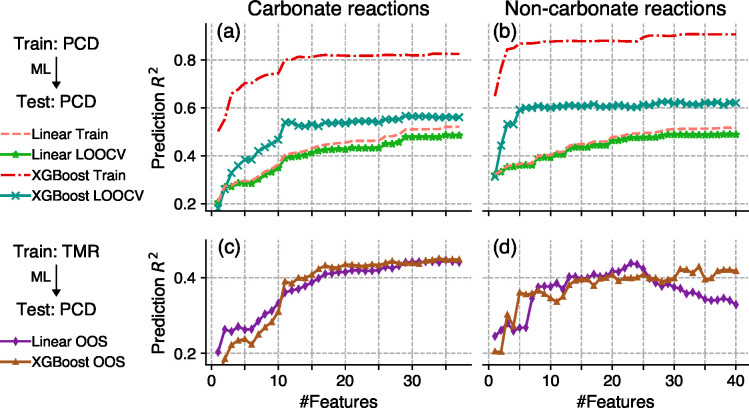
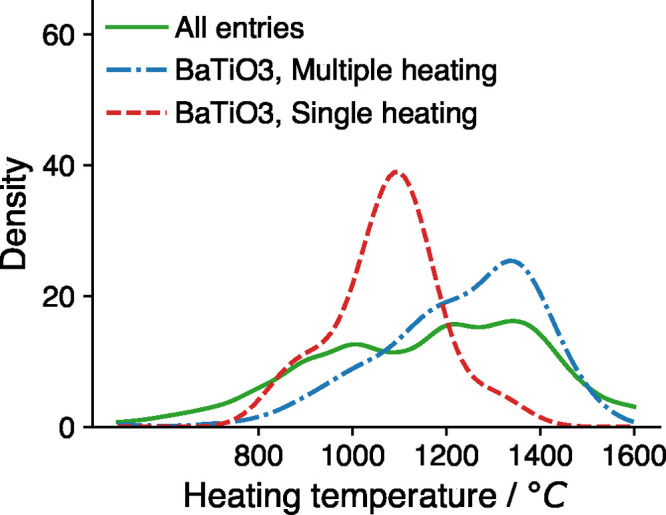
There currently exist no quantitative methods to determine the appropriate conditions for solid-state synthesis. This not only hinders the experimental realization of novel materials but also complicates the interpretation and understanding of solid-state reaction mechanisms. Here, we demonstrate a machine-learning approach that predicts synthesis conditions using large solid-state synthesis data sets text-mined from scientific journal articles. Using feature importance ranking analysis, we discovered that optimal heating temperatures have strong correlations with the stability of precursor materials quantified using melting points and formation energies (Δ , Δ ). In contrast, features derived from the thermodynamics of synthesis-related reactions did not directly correlate to the chosen heating temperatures. This correlation between optimal solid-state heating temperature and precursor stability extends Tamman's rule from intermetallics to oxide systems, suggesting the importance of reaction kinetics in determining synthesis conditions. Heating times are shown to be strongly correlated with the chosen experimental procedures and instrument setups, which may be indicative of human bias in the data set. Using these predictive features, we constructed machine-learning models with good performance and general applicability to predict the conditions required to synthesize diverse chemical systems.
目前还没有定量方法来确定固态合成的合适条件。这不仅阻碍了新型材料的实验实现,也使固态反应机理的解释和理解变得复杂。在这里,我们展示了一种机器学习方法,该方法使用从科学期刊文章中挖掘的大量固态合成数据集来预测合成条件。通过特征重要性排序分析,我们发现最佳加热温度与使用熔点和形成能(Δ ,Δ )量化的前驱体材料稳定性密切相关。相比之下,与合成相关反应的热力学衍生特征与所选加热温度没有直接关联。最佳固态加热温度与前驱体稳定性之间的这种相关性将塔曼规则从金属间化合物扩展到氧化物体系,表明反应动力学在确定合成条件中的重要性。加热时间与所选实验程序和仪器设置密切相关,这可能表明数据集中存在人为偏差。利用这些预测特征,我们构建了具有良好性能和普遍适用性的机器学习模型,以预测合成各种化学体系所需的条件。