Center for Biophysics and Computational Biology, Temple University, Philadelphia, United States.
Department of Chemistry, Temple University, Philadelphia, United States.
Elife. 2022 Dec 23;11:e83368. doi: 10.7554/eLife.83368.
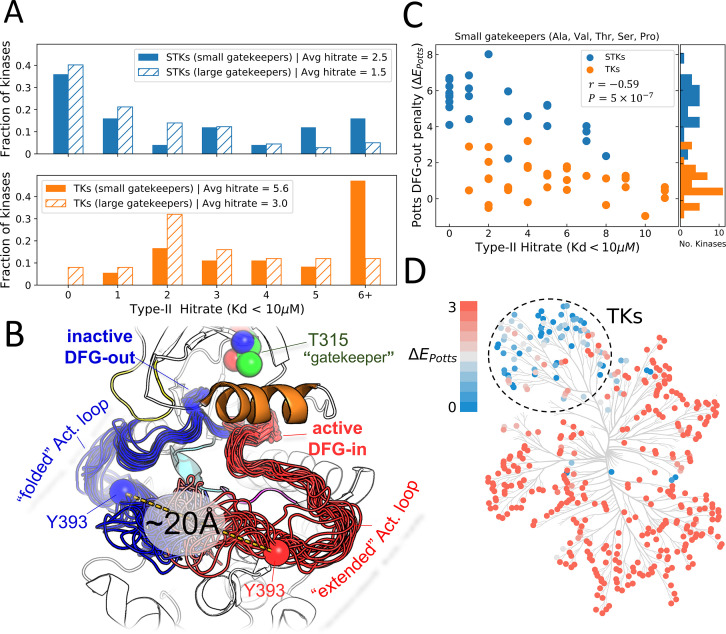
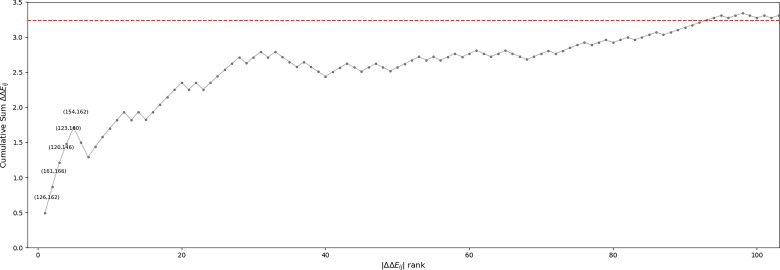
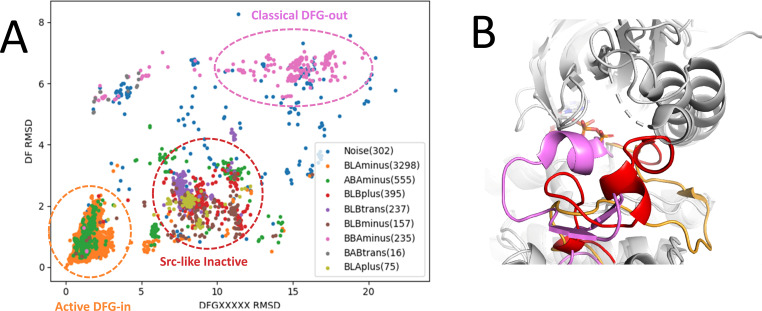
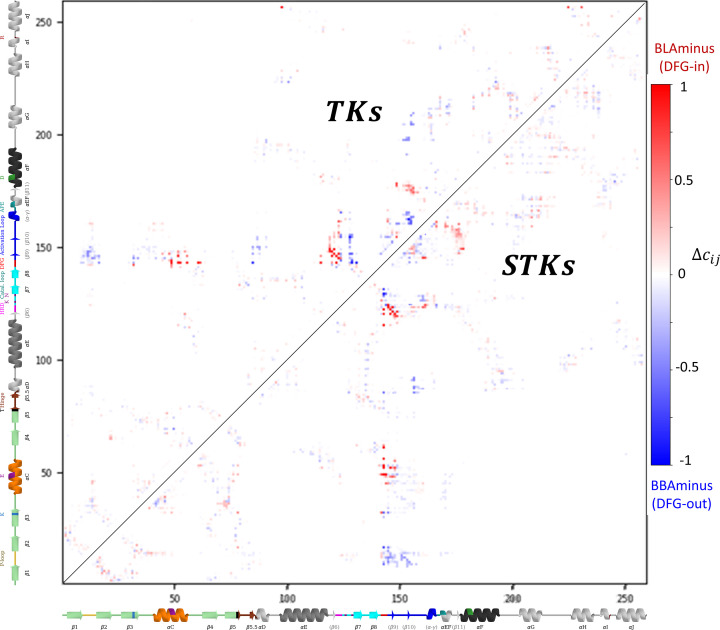
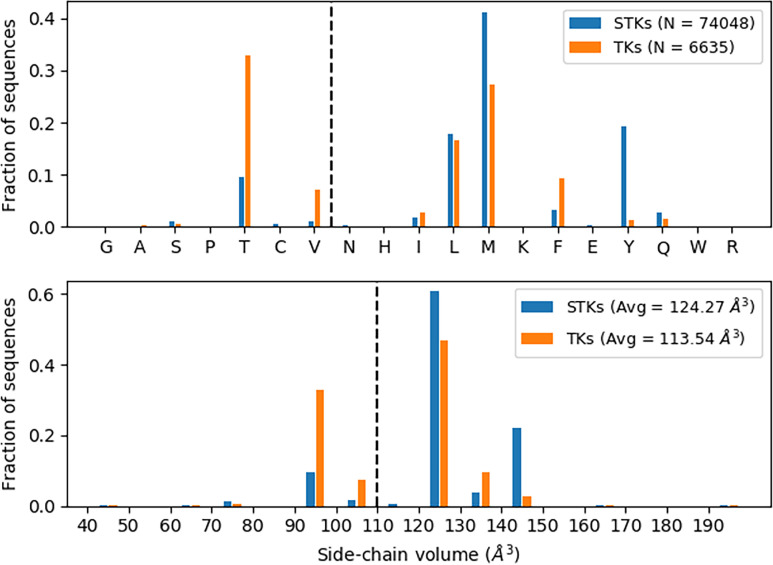
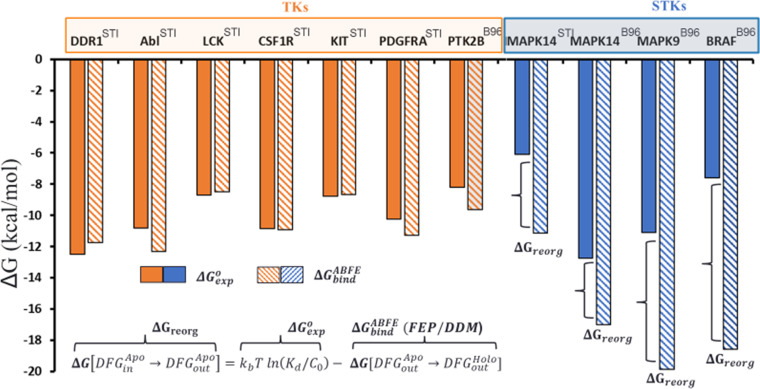
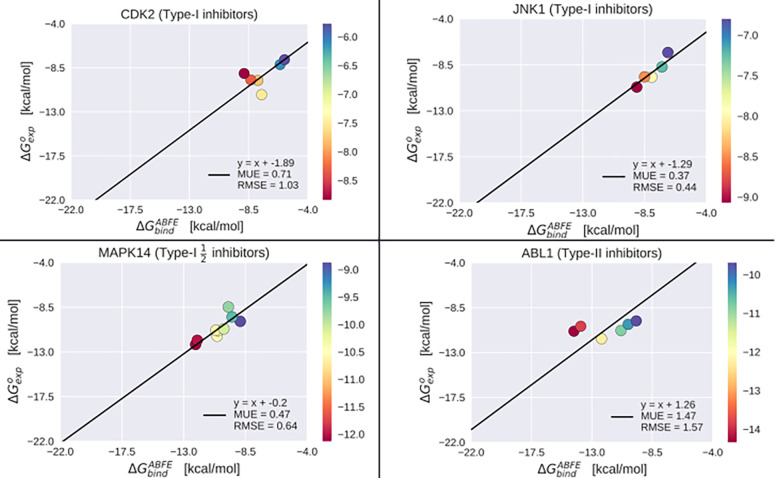
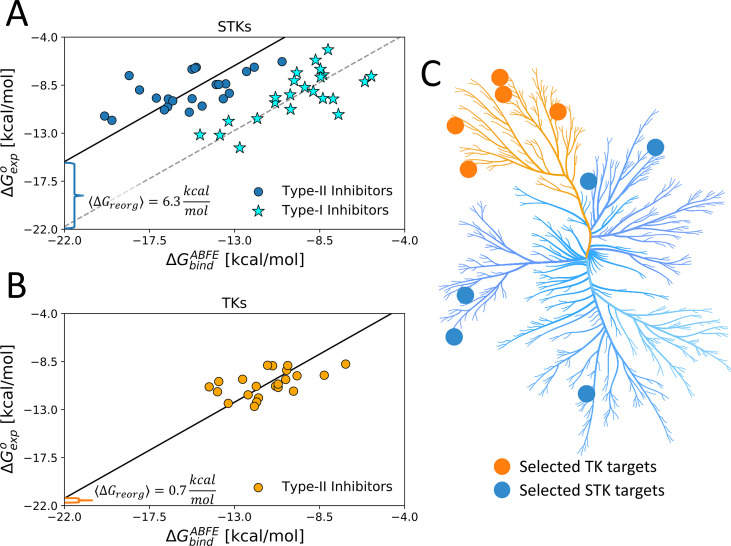
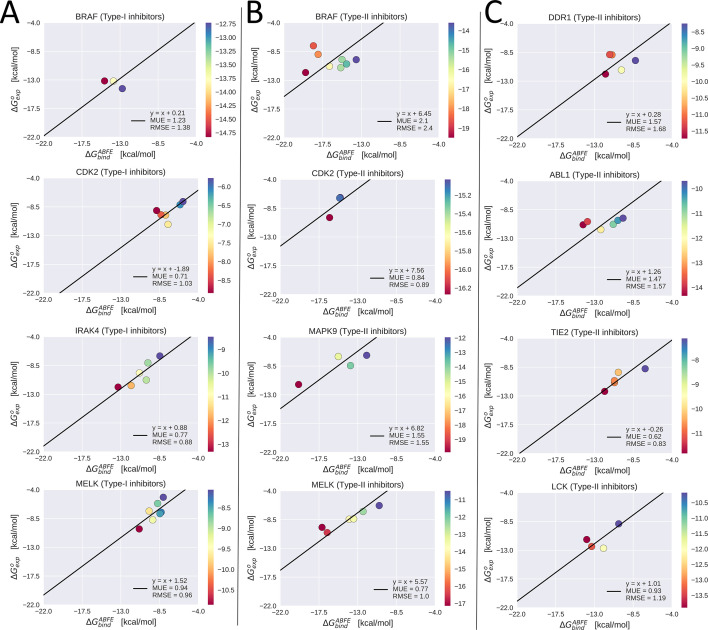
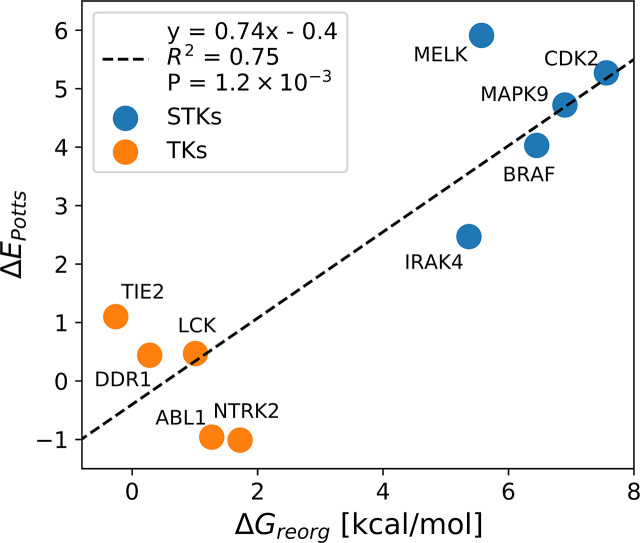
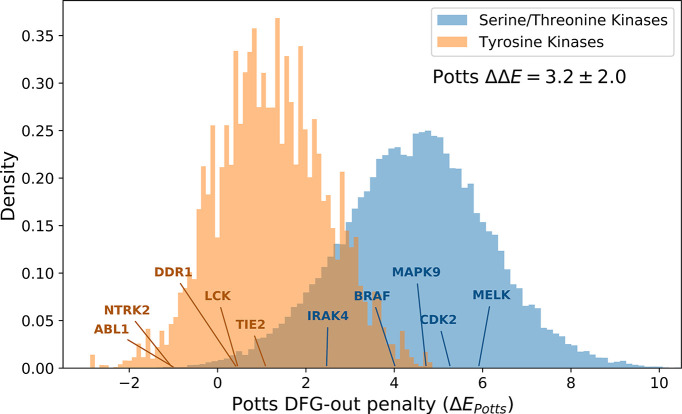
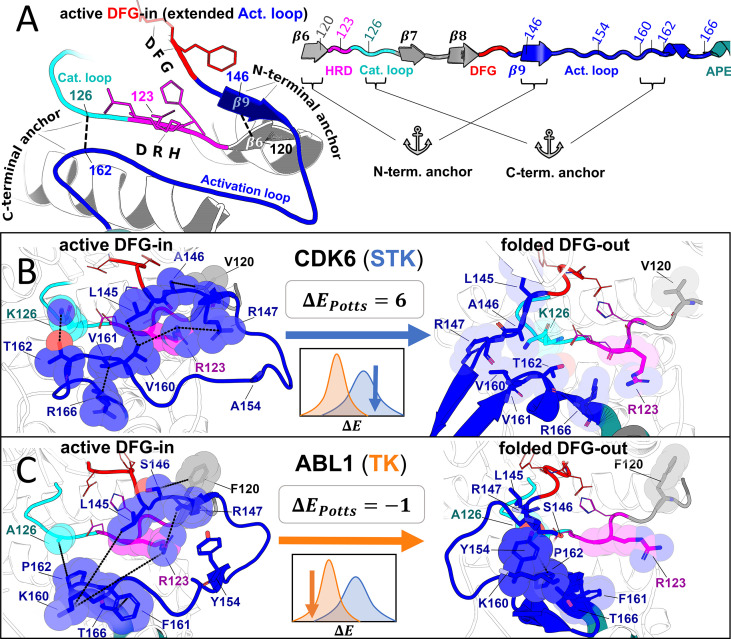
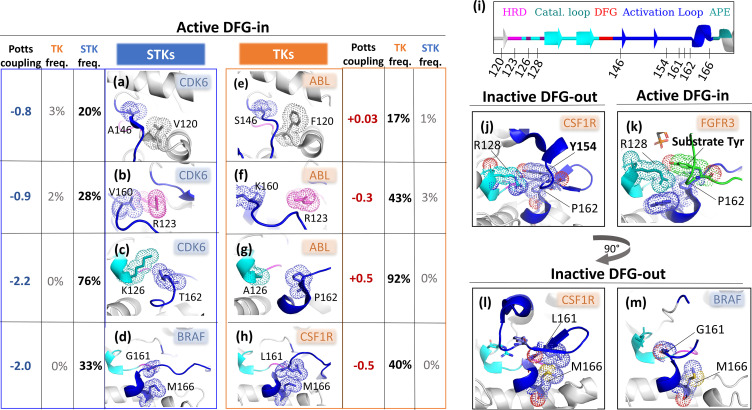
Inactive conformations of protein kinase catalytic domains where the DFG motif has a "DFG-out" orientation and the activation loop is folded present a druggable binding pocket that is targeted by FDA-approved 'type-II inhibitors' in the treatment of cancers. Tyrosine kinases (TKs) typically show strong binding affinity with a wide spectrum of type-II inhibitors while serine/threonine kinases (STKs) usually bind more weakly which we suggest here is due to differences in the folded to extended conformational equilibrium of the activation loop between TKs vs. STKs. To investigate this, we use sequence covariation analysis with a Potts Hamiltonian statistical energy model to guide absolute binding free-energy molecular dynamics simulations of 74 protein-ligand complexes. Using the calculated binding free energies together with experimental values, we estimated free-energy costs for the large-scale (~17-20 Å) conformational change of the activation loop by an indirect approach, circumventing the very challenging problem of simulating the conformational change directly. We also used the Potts statistical potential to thread large sequence ensembles over active and inactive kinase states. The structure-based and sequence-based analyses are consistent; together they suggest TKs evolved to have free-energy penalties for the classical 'folded activation loop' DFG-out conformation relative to the active conformation, that is, on average, 4-6 kcal/mol smaller than the corresponding values for STKs. Potts statistical energy analysis suggests a molecular basis for this observation, wherein the activation loops of TKs are more weakly 'anchored' against the catalytic loop motif in the active conformation and form more stable substrate-mimicking interactions in the inactive conformation. These results provide insights into the molecular basis for the divergent functional properties of TKs and STKs, and have pharmacological implications for the target selectivity of type-II inhibitors.
无活性构象的蛋白激酶催化结构域中,DFG 基序具有“DFG-out”取向,激活环折叠,呈现出可结合的口袋,可被 FDA 批准的“II 型抑制剂”靶向,用于癌症治疗。酪氨酸激酶(TKs)通常与广泛的 II 型抑制剂表现出强烈的结合亲和力,而丝氨酸/苏氨酸激酶(STKs)通常结合较弱,我们在这里认为这是由于 TKs 与 STKs 之间激活环折叠到扩展构象平衡的差异所致。为了研究这一点,我们使用 Potts 哈密顿统计能量模型的序列协变分析来指导 74 个蛋白-配体复合物的绝对结合自由能分子动力学模拟。使用计算的结合自由能与实验值相结合,我们通过间接方法估计了激活环的大规模(~17-20 Å)构象变化的自由能成本,避免了直接模拟构象变化的极具挑战性的问题。我们还使用 Potts 统计势来对活性和非活性激酶状态的大序列集合进行穿线。基于结构的和基于序列的分析是一致的;它们共同表明,TKs 的进化导致了经典的“折叠激活环”DFG-out 构象相对于活性构象的自由能罚值,即平均比 STKs 的相应值小 4-6 kcal/mol。Potts 统计能量分析为这一观察结果提供了分子基础,其中 TKs 的激活环在活性构象中对催化环基序的“锚定”较弱,并在非活性构象中形成更稳定的底物模拟相互作用。这些结果深入了解了 TKs 和 STKs 功能特性差异的分子基础,并对 II 型抑制剂的靶选择性具有药理学意义。