Ramsbottom Kerry A, Prakash Ananth, Riverol Yasset Perez, Camacho Oscar Martin, Sun Zhi, Kundu Deepti J, Bowler-Barnett Emily, Martin Maria, Fan Jun, Chebotarov Dmytro, McNally Kenneth L, Deutsch Eric W, Vizcaíno Juan Antonio, Jones Andrew R
Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, L69 7BE, United Kingdom.
European Molecular Biology Laboratory, EMBL-European Bioinformatics Institute (EMBL-EBI), Hinxton, Cambridge, CB10 1SD, United Kingdom.
bioRxiv. 2023 Nov 17:2023.11.17.567512. doi: 10.1101/2023.11.17.567512.
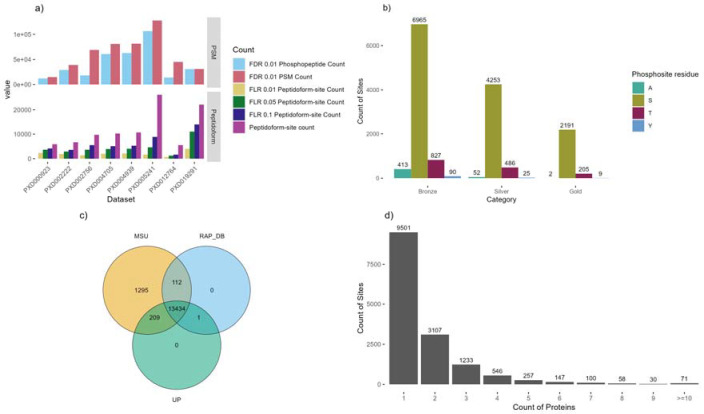
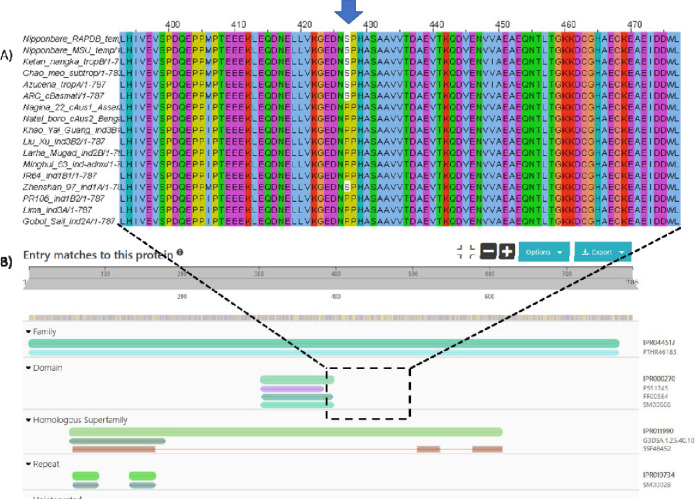
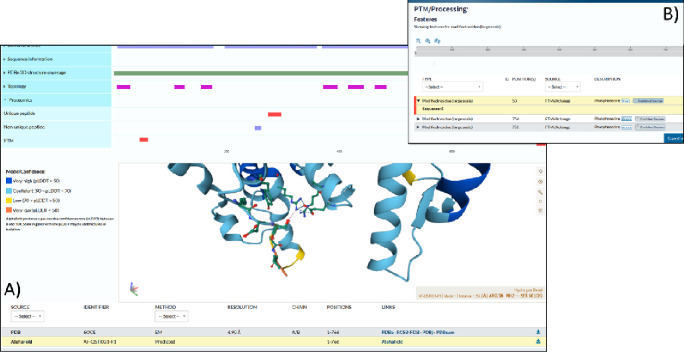
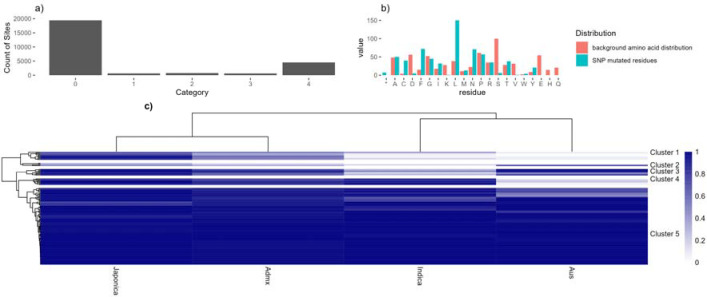
Phosphorylation is the most studied post-translational modification, and has multiple biological functions. In this study, we have re-analysed publicly available mass spectrometry proteomics datasets enriched for phosphopeptides from Asian rice (). In total we identified 15,522 phosphosites on serine, threonine and tyrosine residues on rice proteins. We identified sequence motifs for phosphosites, and link motifs to enrichment of different biological processes, indicating different downstream regulation likely caused by different kinase groups. We cross-referenced phosphosites against the rice 3,000 genomes, to identify single amino acid variations (SAAVs) within or proximal to phosphosites that could cause loss of a site in a given rice variety. The data was clustered to identify groups of sites with similar patterns across rice family groups, for example those highly conserved in Japonica, but mostly absent in Aus type rice varieties - known to have different responses to drought. These resources can assist rice researchers to discover alleles with significantly different functional effects across rice varieties. The data has been loaded into UniProt Knowledge-Base - enabling researchers to visualise sites alongside other data on rice proteins e.g. structural models from AlphaFold2, PeptideAtlas and the PRIDE database - enabling visualisation of source evidence, including scores and supporting mass spectra.
磷酸化是研究最为深入的翻译后修饰,具有多种生物学功能。在本研究中,我们重新分析了公开可用的质谱蛋白质组学数据集,这些数据集富集了来自亚洲水稻的磷酸肽。我们总共在水稻蛋白的丝氨酸、苏氨酸和酪氨酸残基上鉴定出15,522个磷酸化位点。我们确定了磷酸化位点的序列基序,并将基序与不同生物学过程的富集联系起来,表明不同激酶组可能导致不同的下游调控。我们将磷酸化位点与水稻3000基因组进行交叉比对,以识别磷酸化位点内部或附近可能导致特定水稻品种中某个位点缺失的单氨基酸变异(SAAV)。数据被聚类以识别水稻家族组中具有相似模式的位点组,例如在粳稻中高度保守但在Aus型水稻品种中大多不存在的那些位点——已知Aus型水稻品种对干旱有不同反应。这些资源可以帮助水稻研究人员发现不同水稻品种间功能效应显著不同的等位基因。这些数据已被加载到UniProt知识库中,使研究人员能够将位点与水稻蛋白的其他数据(如来自AlphaFold2的结构模型、PeptideAtlas和PRIDE数据库的数据)一起可视化,从而能够可视化来源证据,包括得分和支持性质谱。