Department of Pharmacy, Uppsala University, Uppsala, Sweden.
Division of Quantitative Methods and Modeling, Office of Research Standards, Office of Generic Drugs, Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, Maryland, USA.
CPT Pharmacometrics Syst Pharmacol. 2024 Oct;13(10):1734-1747. doi: 10.1002/psp4.13216. Epub 2024 Aug 23.
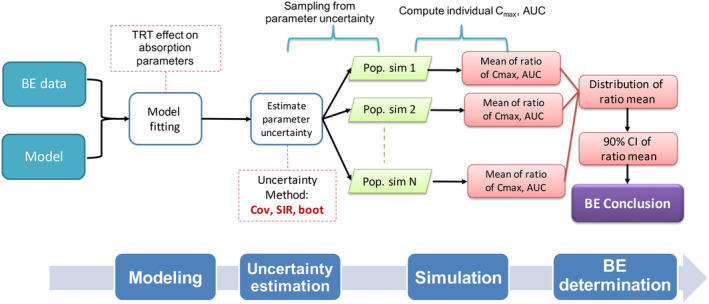
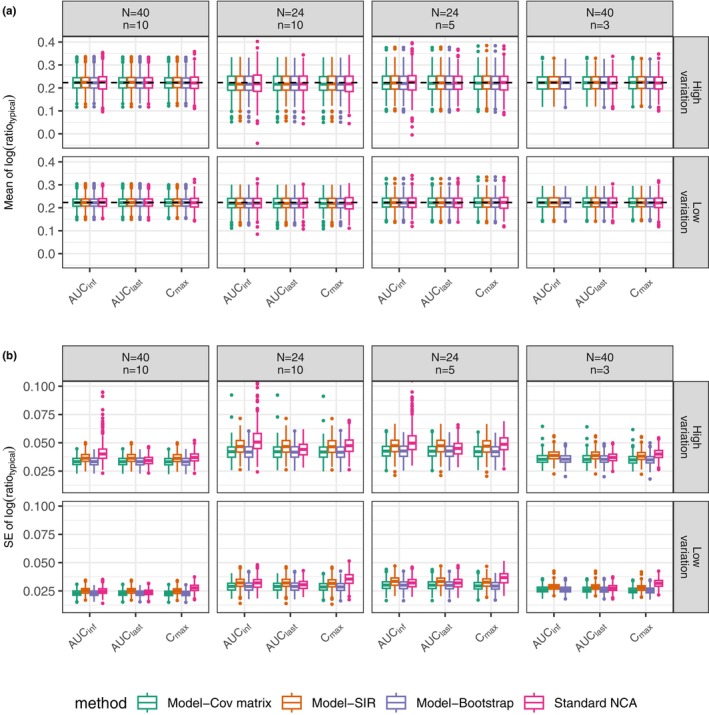
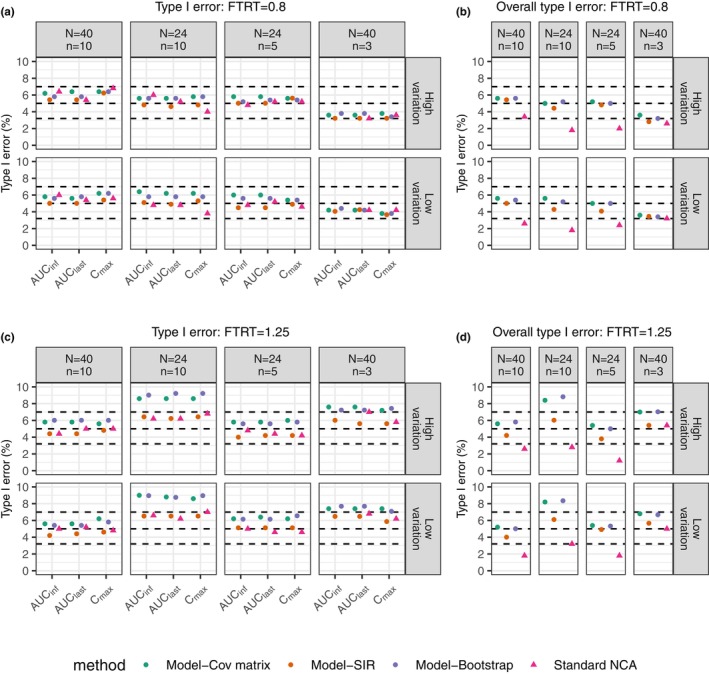
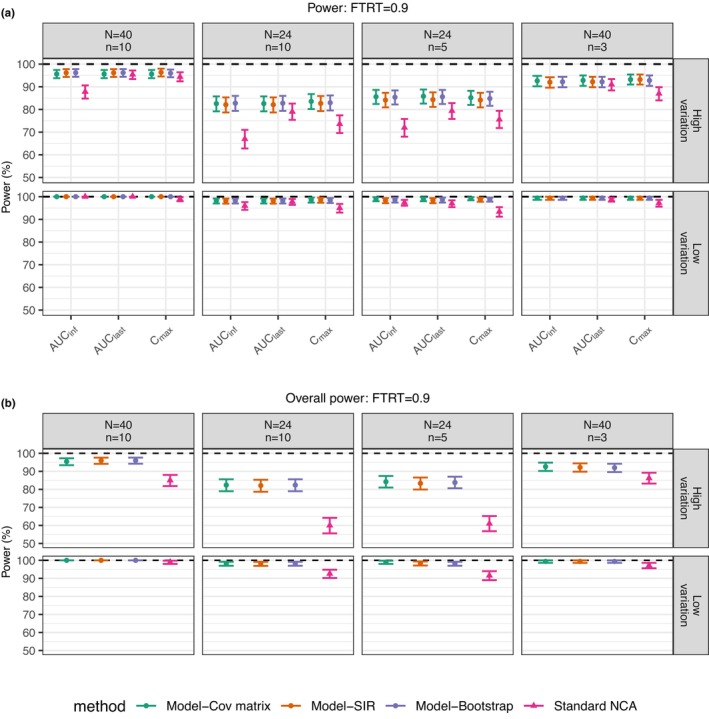
By applying nonlinear mixed-effect (NLME) models, model-integrated evidence (MIE) approaches are able to analyze bioequivalence (BE) data with pharmacokinetic end points that have sparse sampling, which is problematic for non-compartmental analysis (NCA). However, MIE approaches may suffer from inflation of type I error due to underestimation of parameter uncertainty and to the assumption of asymptotic normality. In this study, we developed a MIE BE analysis method that is based on a pre-defined model and consists of several steps including model fitting, uncertainty assessment, simulation, and BE determination. The presented MIE approach has several improvements compared with the previously reported model-integrated methods: (1) treatment, sequence, and period effects are only added to absorption parameters (such as relative bioavailability and rate of absorption) instead of all PK parameters; (2) a simulation step is performed to generate confidence intervals of the pharmacokinetic metrics for BE assessment; and (3) in an effort to maintain type I error, two more advanced parameter uncertainty evaluation approaches are explored, a nonparametric (case resampling) bootstrap, and sampling importance resampling (SIR). To evaluate the developed method and compare the uncertainty assessment methods, simulation experiments were performed for BE studies using a two-way crossover design with different amounts of information (sparse to rich designs) and levels of variability. Based on the simulation results, the method using SIR for parameter uncertainty quantification controls type I error at the nominal level of 0.05 (i.e., the significance level set for BE evaluation) even for studies with small sample size and/or sparse sampling. As expected, our MIE approach for BE assessment exhibited higher power than the NCA-based method, especially as the data becomes sparser and/or more variable.
通过应用非线性混合效应 (NLME) 模型,模型集成证据 (MIE) 方法能够分析具有稀疏采样的药代动力学终点的生物等效性 (BE) 数据,这对于非房室分析 (NCA) 来说是有问题的。然而,由于参数不确定性的低估和渐近正态性的假设,MIE 方法可能会导致 I 型错误的膨胀。在这项研究中,我们开发了一种基于预定义模型的 MIE BE 分析方法,该方法包括模型拟合、不确定性评估、模拟和 BE 确定等几个步骤。与以前报道的模型集成方法相比,提出的 MIE 方法有几个改进:(1) 治疗、序列和周期效应仅添加到吸收参数(如相对生物利用度和吸收速率),而不是所有 PK 参数;(2) 进行模拟步骤以生成用于 BE 评估的药代动力学指标的置信区间;(3) 为了保持 I 型错误,探索了两种更先进的参数不确定性评估方法,即非参数(病例重采样)引导和采样重要性重采样 (SIR)。为了评估所开发的方法并比较不确定性评估方法,我们使用具有不同信息量(从稀疏到丰富设计)和变异性水平的双向交叉设计进行了 BE 研究的模拟实验。基于模拟结果,使用 SIR 进行参数不确定性量化的方法在 0.05 的名义水平(即 BE 评估设定的显著性水平)下控制 I 型错误,即使对于样本量小和/或稀疏采样的研究也是如此。正如预期的那样,我们的 BE 评估 MIE 方法比基于 NCA 的方法表现出更高的功效,尤其是当数据变得更稀疏和/或更具变异性时。