Mosaiques diagnostics and therapeutics, Hannover, Germany.
BMC Bioinformatics. 2010 Dec 10;11:594. doi: 10.1186/1471-2105-11-594.
The purpose of this manuscript is to provide, based on an extensive analysis of a proteomic data set, suggestions for proper statistical analysis for the discovery of sets of clinically relevant biomarkers. As tractable example we define the measurable proteomic differences between apparently healthy adult males and females. We choose urine as body-fluid of interest and CE-MS, a thoroughly validated platform technology, allowing for routine analysis of a large number of samples. The second urine of the morning was collected from apparently healthy male and female volunteers (aged 21-40) in the course of the routine medical check-up before recruitment at the Hannover Medical School.
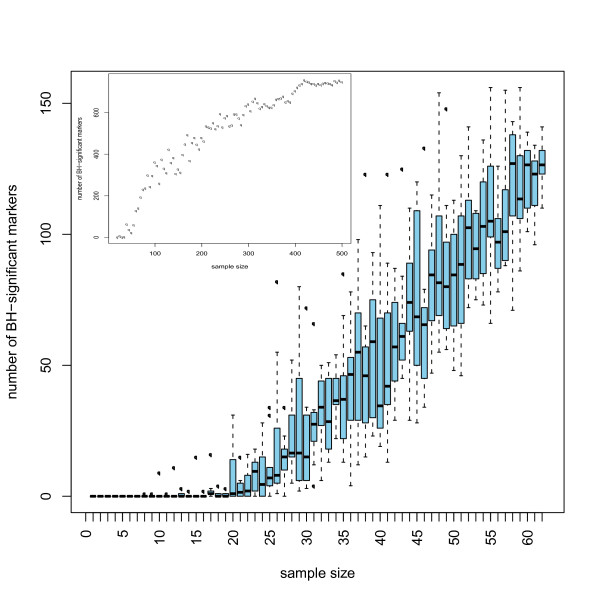
We found that the Wilcoxon-test is best suited for the definition of potential biomarkers. Adjustment for multiple testing is necessary. Sample size estimation can be performed based on a small number of observations via resampling from pilot data. Machine learning algorithms appear ideally suited to generate classifiers. Assessment of any results in an independent test-set is essential.
Valid proteomic biomarkers for diagnosis and prognosis only can be defined by applying proper statistical data mining procedures. In particular, a justification of the sample size should be part of the study design.
本文旨在基于对蛋白质组数据集的广泛分析,为发现具有临床相关性的生物标志物集提供合适的统计分析建议。作为一个可行的例子,我们定义了在明显健康的成年男性和女性之间可测量的蛋白质组差异。我们选择尿液作为感兴趣的体液,并选择 CE-MS 作为经过充分验证的平台技术,可用于常规分析大量样本。在汉诺威医学院招募前,在常规体检过程中收集了来自明显健康的男性和女性志愿者(年龄在 21-40 岁之间)的第二次晨尿。
我们发现 Wilcoxon 检验最适合定义潜在的生物标志物。需要进行多重检验调整。可以通过从试点数据中重新采样来对小样本量进行样本量估计。机器学习算法似乎非常适合生成分类器。在独立测试集中评估任何结果至关重要。
只有通过应用适当的统计数据挖掘程序,才能定义有效的蛋白质组生物标志物用于诊断和预后。特别是,应将样本量的合理性作为研究设计的一部分。