Department of Physiology and Biophysics, University of Washington, Seattle, WA 98195, USA.
J Gen Physiol. 2013 May;141(5):537-55. doi: 10.1085/jgp.201210887.
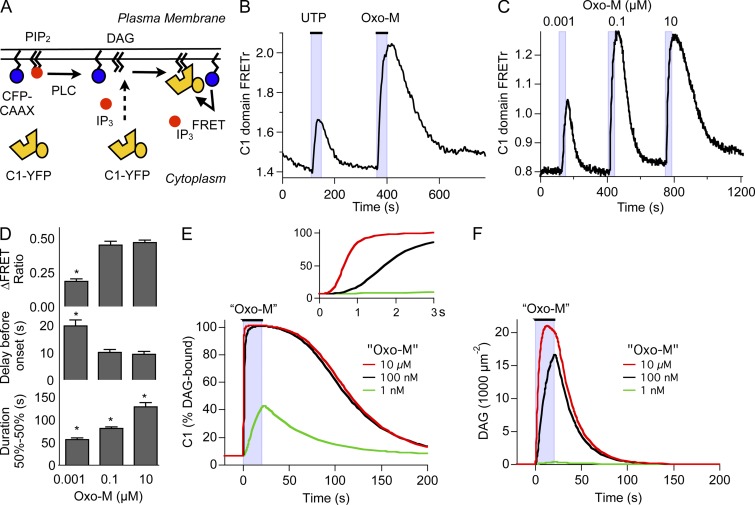
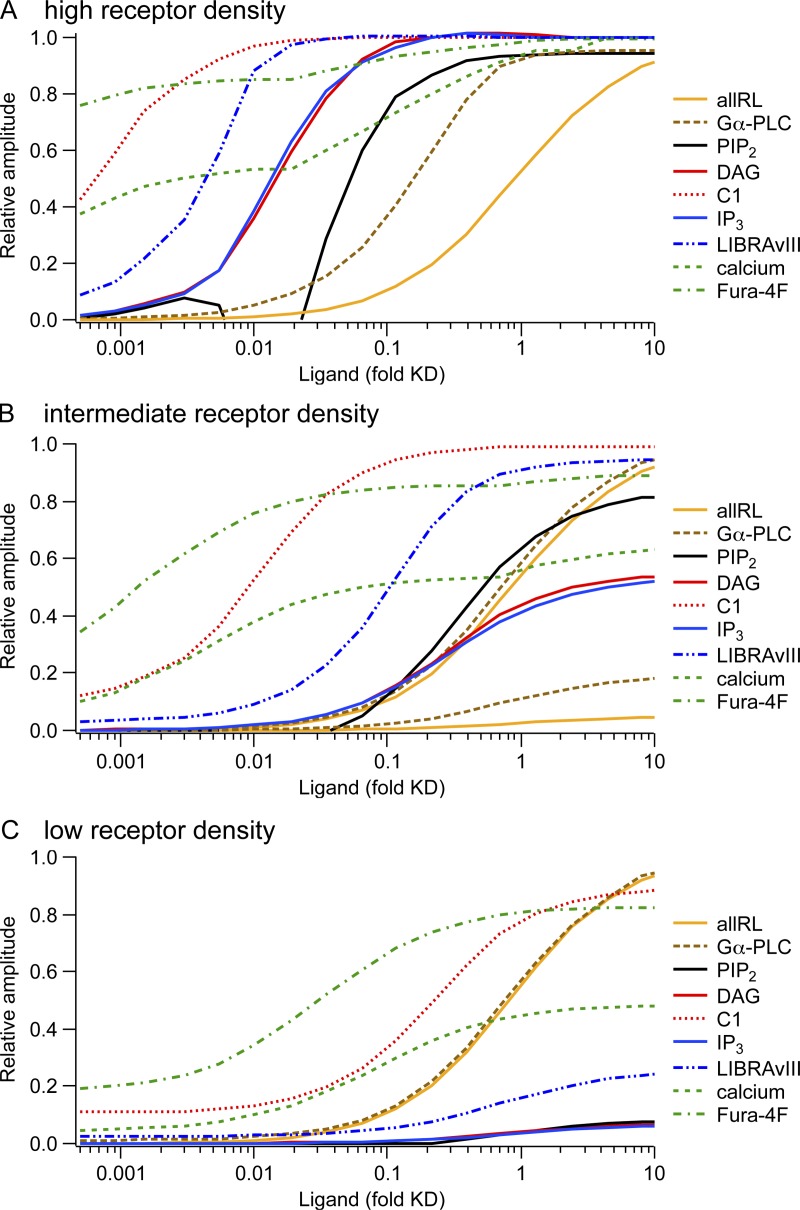
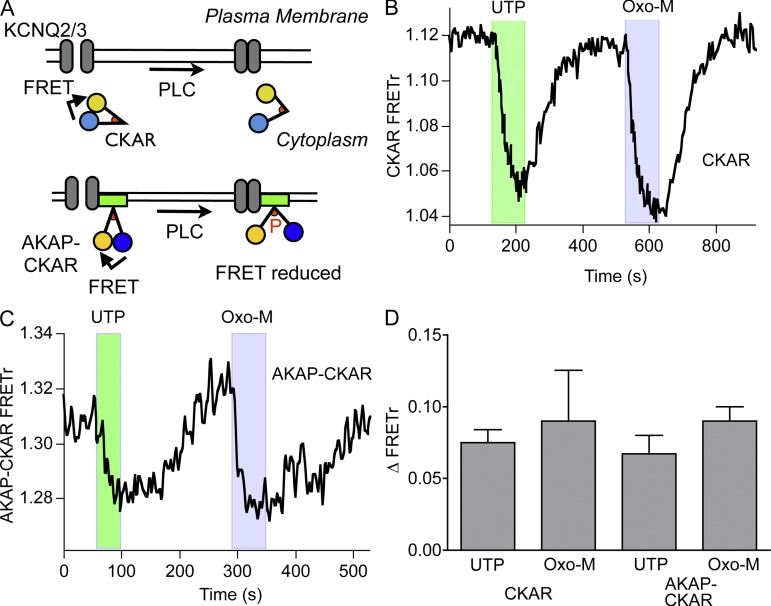
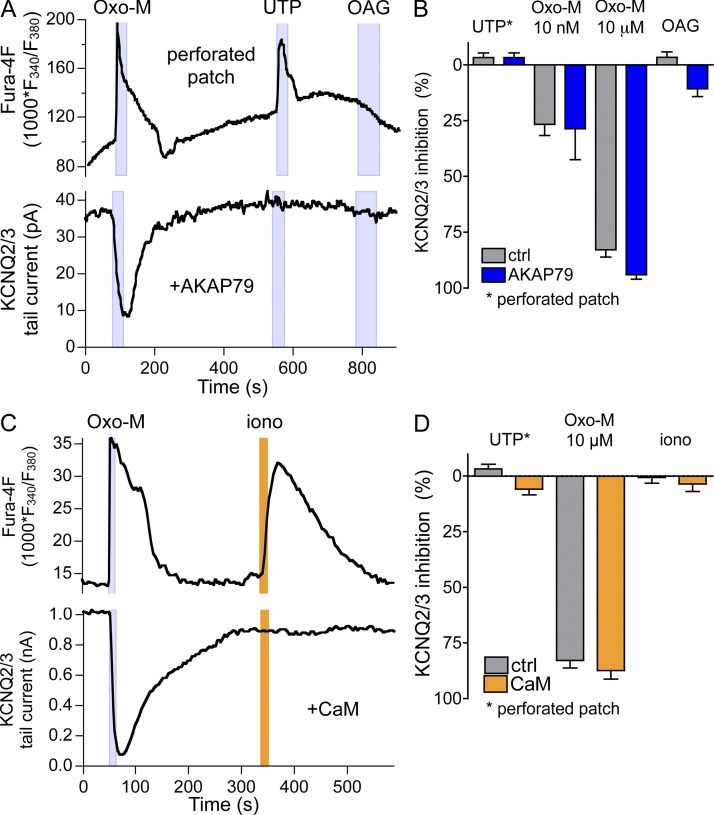
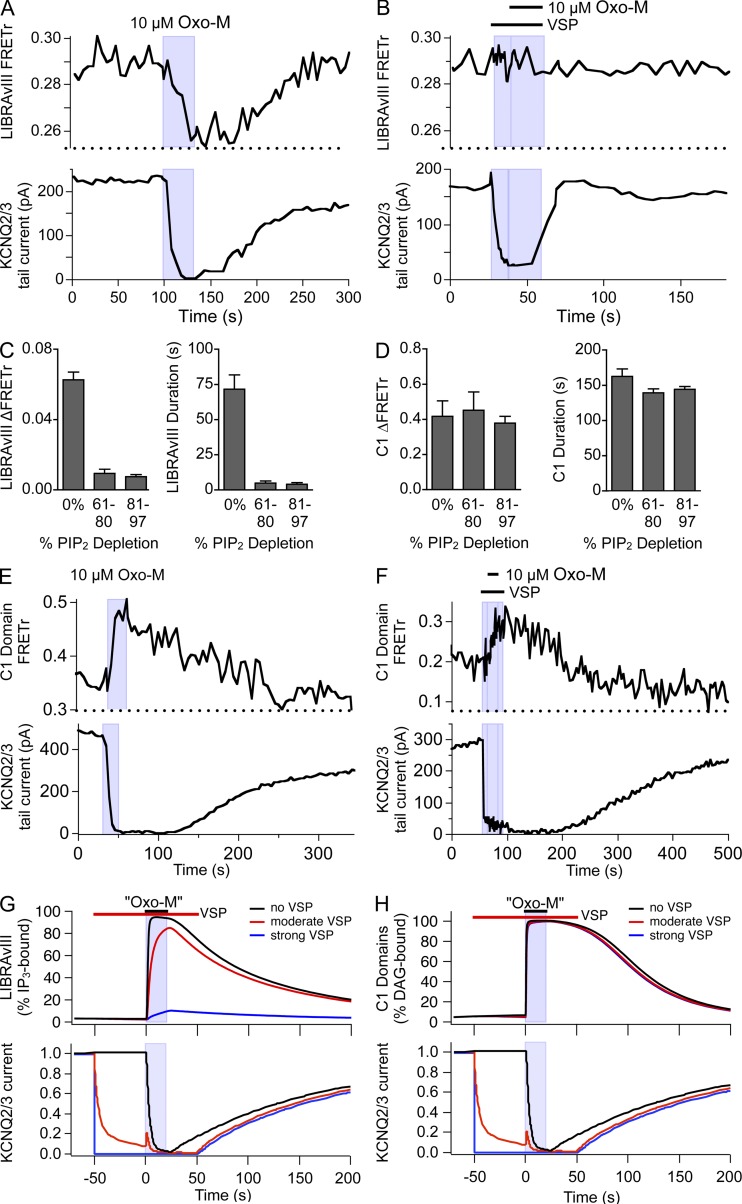
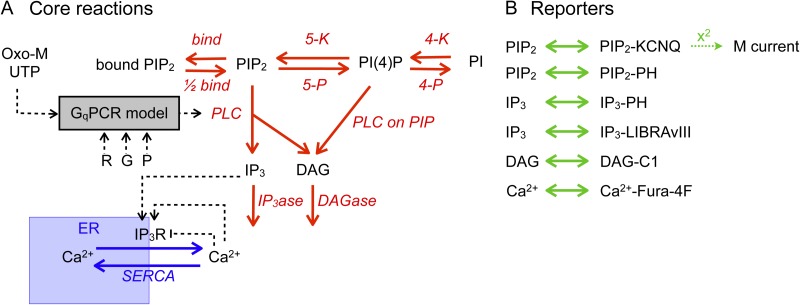
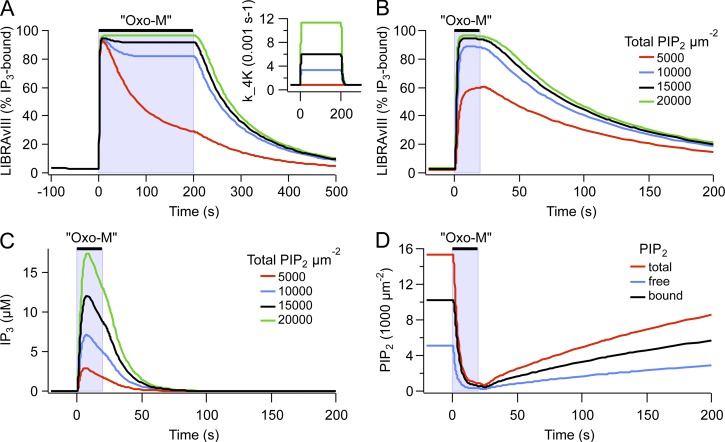
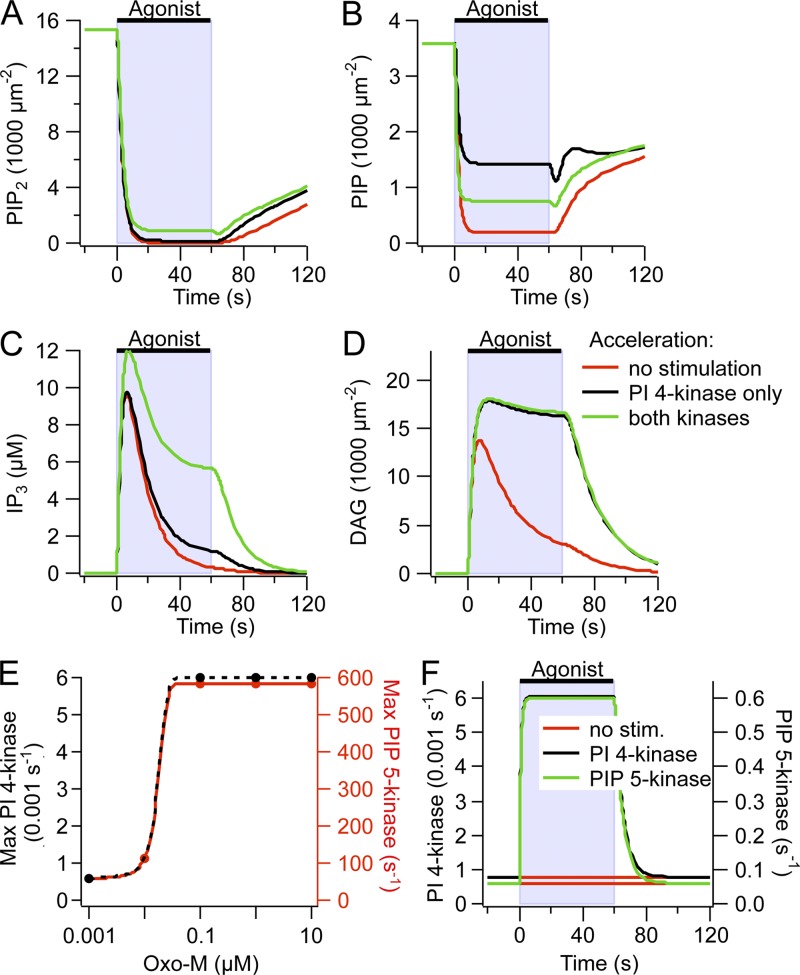
Gq protein-coupled receptors (GqPCRs) of the plasma membrane activate the phospholipase C (PLC) signaling cascade. PLC cleaves the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) into the second messengers diacylgycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), leading to calcium release, protein kinase C (PKC) activation, and in some cases, PIP2 depletion. We determine the kinetics of each of these downstream endpoints and also ask which is responsible for the inhibition of KCNQ2/3 (KV7.2/7.3) potassium channels in single living tsA-201 cells. We measure DAG production and PKC activity by Förster resonance energy transfer-based sensors, and PIP2 by KCNQ2/3 channels. Fully activating endogenous purinergic receptors by uridine 5'triphosphate (UTP) leads to calcium release, DAG production, and PKC activation, but no net PIP2 depletion. Fully activating high-density transfected muscarinic receptors (M1Rs) by oxotremorine-M (Oxo-M) leads to similar calcium, DAG, and PKC signals, but PIP2 is depleted. KCNQ2/3 channels are inhibited by the Oxo-M treatment (85%) and not by UTP (<1%), indicating that depletion of PIP2 is required to inhibit KCNQ2/3 in response to receptor activation. Overexpression of A kinase-anchoring protein (AKAP)79 or calmodulin (CaM) does not increase KCNQ2/3 inhibition by UTP. From these results and measurements of IP3 and calcium presented in our companion paper (Dickson et al. 2013. J. Gen. Physiol. http://dx.doi.org/10.1085/jgp.201210886), we extend our kinetic model for signaling from M1Rs to DAG/PKC and IP3/calcium signaling. We conclude that calcium/CaM and PKC-mediated phosphorylation do not underlie dynamic KCNQ2/3 channel inhibition during GqPCR activation in tsA-201 cells. Finally, our experimental data provide indirect evidence for cleavage of PI(4)P by PLC in living cells, and our modeling revisits/explains the concept of receptor reserve with measurements from all steps of GqPCR signaling.
Gq 蛋白偶联受体(GqPCRs)位于细胞膜上,可激活磷脂酶 C(PLC)信号级联反应。PLC 将膜脂质磷脂酰肌醇 4,5-二磷酸(PIP2)切割成第二信使二酰基甘油(DAG)和肌醇 1,4,5-三磷酸(IP3),导致钙释放、蛋白激酶 C(PKC)激活,在某些情况下还会导致 PIP2 耗竭。我们确定了这些下游终点的每个反应的动力学,还询问了哪种反应负责抑制单个活 tsA-201 细胞中的 KCNQ2/3(KV7.2/7.3)钾通道。我们通过基于Förster 共振能量转移的传感器测量 DAG 产生和 PKC 活性,并通过 KCNQ2/3 通道测量 PIP2。用尿苷 5'-三磷酸(UTP)完全激活内源性嘌呤能受体可导致钙释放、DAG 产生和 PKC 激活,但不会导致净 PIP2 耗竭。用氧震颤素-M(Oxo-M)完全激活高密度转染的毒蕈碱受体(M1R)可导致类似的钙、DAG 和 PKC 信号,但 PIP2 被耗尽。Oxo-M 处理可抑制 KCNQ2/3 通道(85%),而 UTP 处理则不能抑制(<1%),这表明 PIP2 的耗竭是响应受体激活抑制 KCNQ2/3 所必需的。过表达蛋白激酶 A 锚定蛋白(AKAP)79 或钙调蛋白(CaM)不会增加 UTP 对 KCNQ2/3 的抑制作用。从这些结果以及我们的相关论文(Dickson 等人,2013. J. Gen. Physiol. http://dx.doi.org/10.1085/jgp.201210886)中的 IP3 和钙的测量结果,我们扩展了我们的 M1R 信号从 DAG/PKC 和 IP3/钙信号的动力学模型。我们的结论是,在 tsA-201 细胞中 GqPCR 激活期间,钙/CaM 和 PKC 介导的磷酸化不是动态 KCNQ2/3 通道抑制的基础。最后,我们的实验数据提供了 PLC 在活细胞中切割 PI(4)P 的间接证据,我们的模型通过从 GqPCR 信号的所有步骤进行测量,重新审视/解释了受体储备的概念。