Martini Paolo, Sales Gabriele, Brugiolo Mattia, Gandaglia Alessandro, Naso Filippo, De Pittà Cristiano, Spina Michele, Gerosa Gino, Chemello Francesco, Romualdi Chiara, Cagnin Stefano, Lanfranchi Gerolamo
Department of Biology, University of Padova, Padova, Italy; CRIBI Biotechnology Centre, University of Padova, Padova, Italy.
Department of Biology, University of Padova, Padova, Italy.
PLoS One. 2014 Apr 3;9(4):e89755. doi: 10.1371/journal.pone.0089755. eCollection 2014.
Despite the economic and medical importance of the pig, knowledge about its genome organization, gene expression regulation, and molecular mechanisms involved in physiological processes is far from that achieved for mouse and rat, the two most used model organisms in biomedical research. MicroRNAs (miRNAs) are a wide class of molecules that exert a recognized role in gene expression modulation, but only 280 miRNAs in pig have been characterized to date.
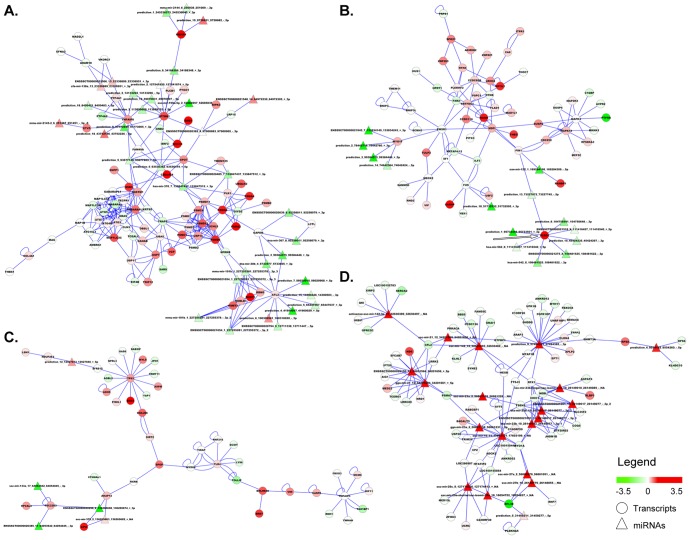
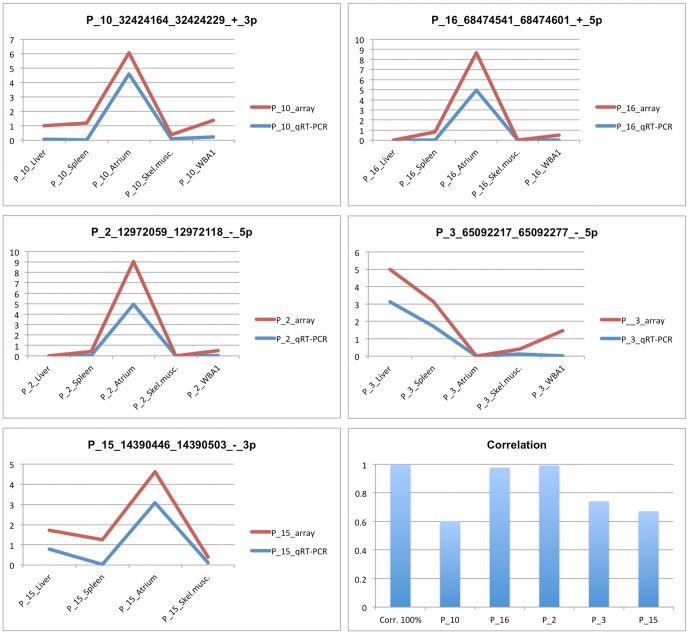
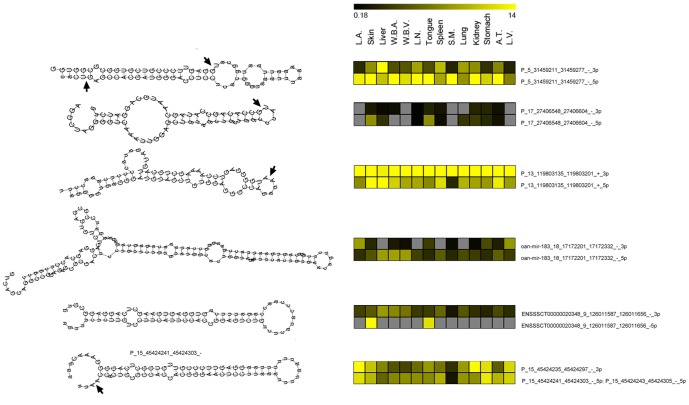
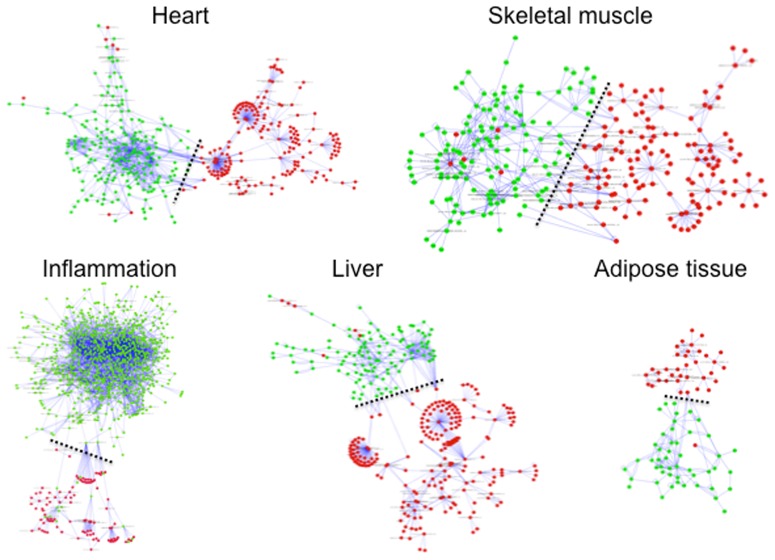
We applied a novel computational approach to predict species-specific and conserved miRNAs in the pig genome, which were then subjected to experimental validation. We experimentally identified candidate miRNAs sequences grouped in high-confidence (424) and medium-confidence (353) miRNAs according to RNA-seq results. A group of miRNAs was also validated by PCR experiments. We established the subtle variability in expression of isomiRs and miRNA-miRNA star couples supporting a biological function for these molecules. Finally, miRNA and mRNA expression profiles produced from the same sample of 20 different tissue of the animal were combined, using a correlation threshold to filter miRNA-target predictions, to identify tissue-specific regulatory networks.
Our data represent a significant progress in the current understanding of miRNAome in pig. The identification of miRNAs, their target mRNAs, and the construction of regulatory circuits will provide new insights into the complex biological networks in several tissues of this important animal model.
尽管猪在经济和医学方面具有重要性,但相较于生物医学研究中最常用的两种模式生物小鼠和大鼠,我们对猪的基因组组织、基因表达调控以及生理过程中涉及的分子机制的了解仍十分有限。微小RNA(miRNA)是一类在基因表达调控中发挥公认作用的分子,但迄今为止,猪中仅鉴定出280种miRNA。
我们应用了一种新颖的计算方法来预测猪基因组中物种特异性和保守的miRNA,随后对其进行实验验证。根据RNA测序结果,我们通过实验鉴定出了分为高可信度(424个)和中等可信度(353个)miRNA的候选miRNA序列。一组miRNA也通过PCR实验得到了验证。我们确定了异源miRNA和miRNA-miRNA星型配对表达中的细微差异,支持了这些分子的生物学功能。最后,将来自动物20种不同组织的同一样本产生的miRNA和mRNA表达谱相结合,使用相关性阈值筛选miRNA靶标预测,以识别组织特异性调控网络。
我们的数据代表了目前对猪miRNA组理解的重大进展。miRNA及其靶标mRNA的鉴定以及调控回路的构建将为这个重要动物模型的多个组织中的复杂生物网络提供新的见解。