Chair of Statistical Bioinformatics, University of Regensburg, Am BioPark 9, 93053, Regensburg, Germany.
Institute of Clinical Microbiology and Hygiene, University Medical Centre, Franz-Josef-Strauß-Allee 11, 93053, Regensburg, Germany.
Microbiome. 2016 Jun 21;4(1):28. doi: 10.1186/s40168-016-0175-0.
Next-generation 16S ribosomal RNA gene sequencing is widely used to determine the relative composition of the mammalian gut microbiomes. However, in the absence of a reference, this does not reveal alterations in absolute abundance of specific operational taxonomic units if microbial loads vary across specimens.
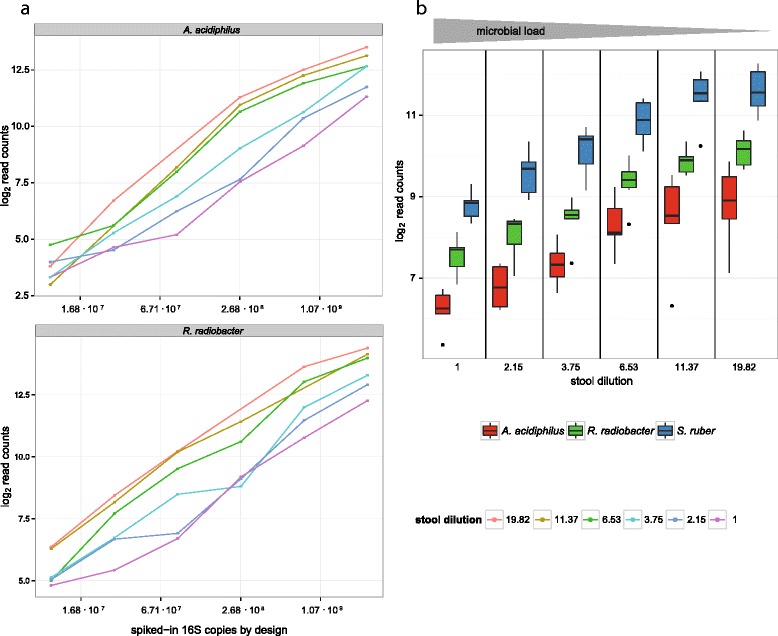
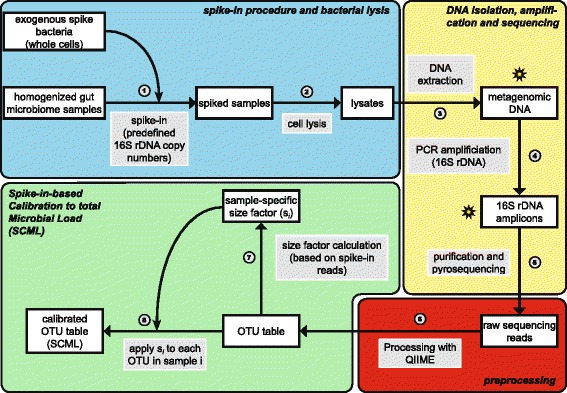
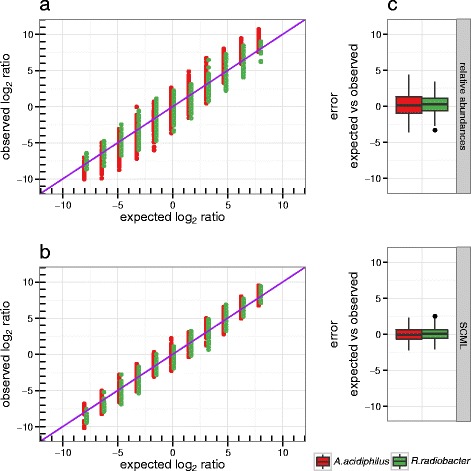
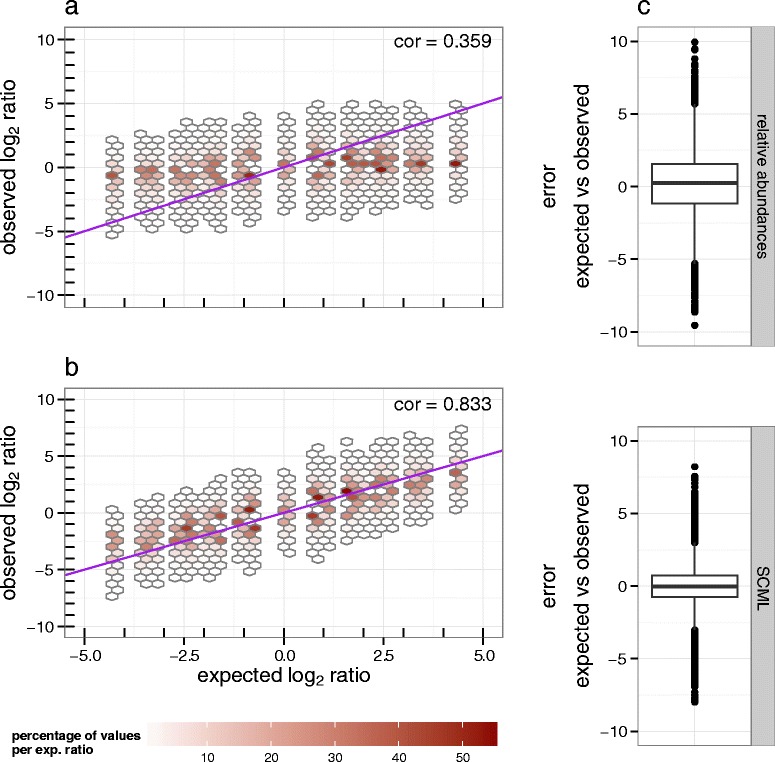
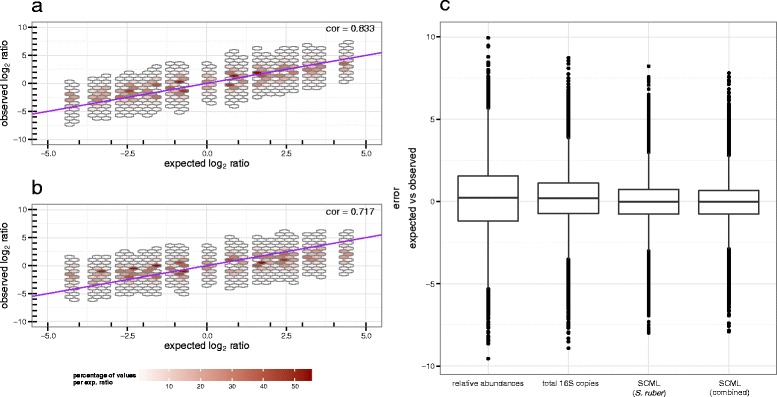
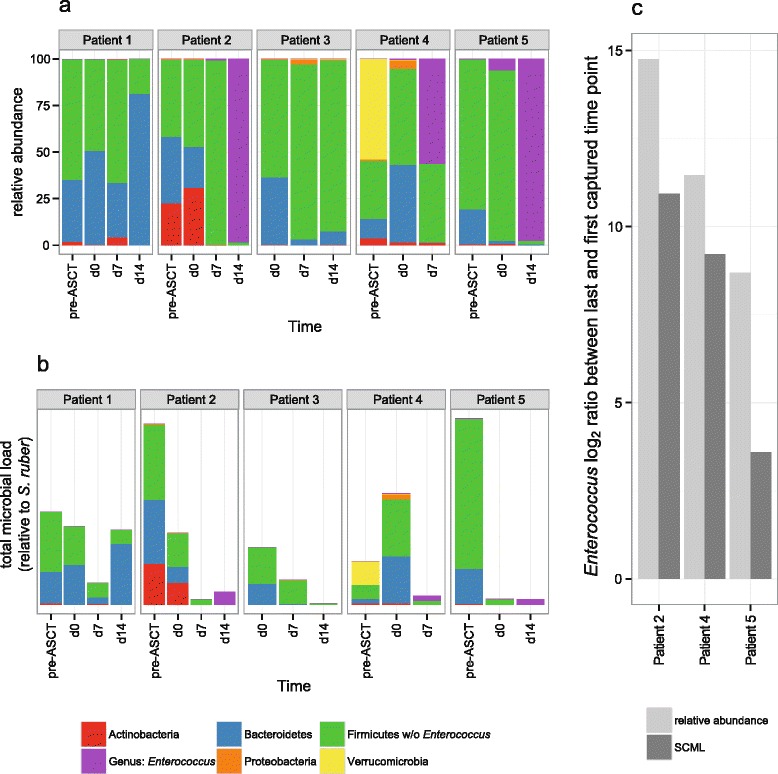
Here we suggest the spiking of exogenous bacteria into crude specimens to quantify ratios of absolute bacterial abundances. We use the 16S rDNA read counts of the spike-in bacteria to adjust the read counts of endogenous bacteria for changes in total microbial loads. Using a series of dilutions of pooled faecal samples from mice containing defined amounts of the spike-in bacteria Salinibacter ruber, Rhizobium radiobacter and Alicyclobacillus acidiphilus, we demonstrate that spike-in-based calibration to microbial loads allows accurate estimation of ratios of absolute endogenous bacteria abundances. Applied to stool specimens of patients undergoing allogeneic stem cell transplantation, we were able to determine changes in both relative and absolute abundances of various phyla, especially the genus Enterococcus, in response to antibiotic treatment and radio-chemotherapeutic conditioning.
Exogenous spike-in bacteria in gut microbiome studies enable estimation of ratios of absolute OTU abundances, providing novel insights into the structure and the dynamics of intestinal microbiomes.
下一代 16S 核糖体 RNA 基因测序被广泛用于确定哺乳动物肠道微生物组的相对组成。然而,如果微生物负荷在标本之间存在差异,由于缺乏参考,这并不能揭示特定操作分类单位的绝对丰度变化。
在这里,我们建议向粗制标本中添加外源细菌,以定量绝对细菌丰度的比值。我们使用外源性细菌的 16S rDNA 读计数来调整内源性细菌的读计数,以适应总微生物负荷的变化。使用一系列含有已知量外源性细菌(盐杆菌 Ruber、放射杆菌 Radiobacter 和嗜酸脂环酸芽孢杆菌 Acidiphilus)的混合粪便样本稀释,我们证明基于外源性细菌的微生物负荷校准可以准确估计绝对内源性细菌丰度的比值。将其应用于接受同种异体干细胞移植的患者的粪便标本中,我们能够确定抗生素治疗和放射化学治疗条件下各种门、属水平的相对和绝对丰度的变化,尤其是肠球菌属。
肠道微生物组研究中外源添加的细菌可用于估计绝对 OTU 丰度的比值,为肠道微生物组的结构和动态提供新的见解。