Kolb Brian, Lentz Levi C, Kolpak Alexie M
Massachusetts Institute of Technology, Mechanical Engineering, Cambridge, MA, 02139, USA.
University of New Mexico, Department of Chemistry and Chemical Biology, Albuquerque, NM, 87110, Mexico.
Sci Rep. 2017 Apr 26;7(1):1192. doi: 10.1038/s41598-017-01251-z.
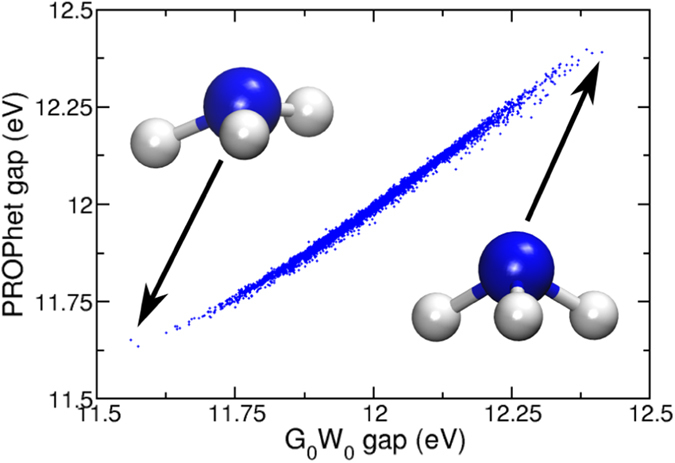
Modern ab initio methods have rapidly increased our understanding of solid state materials properties, chemical reactions, and the quantum interactions between atoms. However, poor scaling often renders direct ab initio calculations intractable for large or complex systems. There are two obvious avenues through which to remedy this problem: (i) develop new, less expensive methods to calculate system properties, or (ii) make existing methods faster. This paper describes an open source framework designed to pursue both of these avenues. PROPhet (short for PROPerty Prophet) utilizes machine learning techniques to find complex, non-linear mappings between sets of material or system properties. The result is a single code capable of learning analytical potentials, non-linear density functionals, and other structure-property or property-property relationships. These capabilities enable highly accurate mesoscopic simulations, facilitate computation of expensive properties, and enable the development of predictive models for systematic materials design and optimization. This work explores the coupling of machine learning to ab initio methods through means both familiar (e.g., the creation of various potentials and energy functionals) and less familiar (e.g., the creation of density functionals for arbitrary properties), serving both to demonstrate PROPhet's ability to create exciting post-processing analysis tools and to open the door to improving ab initio methods themselves with these powerful machine learning techniques.
现代从头算方法迅速增进了我们对固态材料特性、化学反应以及原子间量子相互作用的理解。然而,较差的计算规模扩展性常常使得对于大型或复杂系统的直接从头算计算变得难以处理。有两条明显的途径可以解决这个问题:(i)开发新的、成本更低的方法来计算系统特性,或者(ii)提高现有方法的计算速度。本文描述了一个旨在探索这两条途径的开源框架。PROPhet(PROPerty Prophet的缩写)利用机器学习技术来寻找材料或系统特性集之间复杂的非线性映射。结果是得到一个能够学习解析势、非线性密度泛函以及其他结构-特性或特性-特性关系的单一代码。这些能力使得高精度的介观模拟成为可能,便于计算昂贵的特性,并能够开发用于系统材料设计和优化的预测模型。这项工作通过熟悉的方式(例如创建各种势和能量泛函)以及不太熟悉的方式(例如为任意特性创建密度泛函)探索了机器学习与从头算方法的耦合,既展示了PROPhet创建令人兴奋的后处理分析工具的能力,也为利用这些强大的机器学习技术改进从头算方法本身打开了大门。