Russo Edda, Bacci Giovanni, Chiellini Carolina, Fagorzi Camilla, Niccolai Elena, Taddei Antonio, Ricci Federica, Ringressi Maria N, Borrelli Rossella, Melli Filippo, Miloeva Manouela, Bechi Paolo, Mengoni Alessio, Fani Renato, Amedei Amedeo
Immunology, Department of Clinical and Experimental Medicine, University of Florence, Florence, Italy.
Department of Biology, University of Florence, Florence, Italy.
Front Microbiol. 2018 Jan 12;8:2699. doi: 10.3389/fmicb.2017.02699. eCollection 2017.
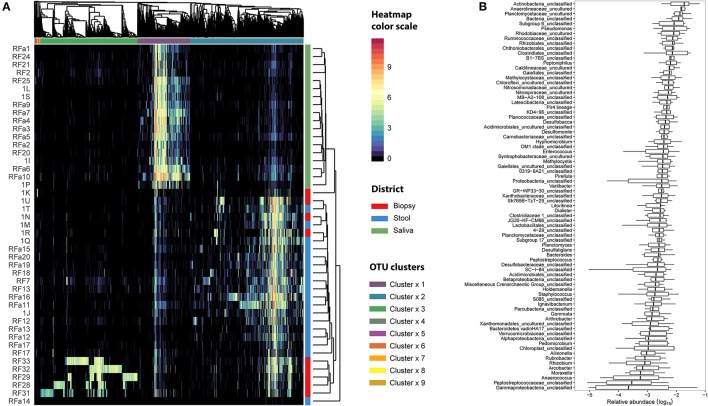
In this study Next-Generation Sequencing (NGS) was used to analyze and compare human microbiota from three different compartments, i.e., saliva, feces, and cancer tissue (CT), of a selected cohort of 10 Italian patients with colorectal cancer (CRC) vs. 10 healthy controls (saliva and feces). Furthermore, the abundance in the same body site was investigated through real-time quantitative polymerase chain reaction (qPCR) to assess the association with CRC. Differences in bacterial composition, abundance in healthy controls vs. CRC patients, and the association of with clinical parameters were observed. Taxonomic analysis based on 16S rRNA gene, revealed the presence of three main bacterial phyla, which includes about 80% of reads: (39.18%), (30.36%), and (10.65%). The results highlighted the presence of different bacterial compositions; in particular, the fecal samples of CRC patients seemed to be enriched with , whereas in the fecal samples of healthy controls were one of the major phyla detected though these differences were not statistically significant. The CT samples showed the highest alpha diversity values. These results emphasize a different taxonomic composition of feces from CRC compared to healthy controls. Despite the low number of samples included in the study, these results suggest the importance of microbiota in the CRC progression and could pave the way to the development of therapeutic interventions and novel microbial-related diagnostic tools in CRC patients.
在本研究中,采用新一代测序(NGS)技术对10名意大利结直肠癌(CRC)患者与10名健康对照者(唾液和粪便)组成的选定队列的三个不同部位,即唾液、粪便和癌组织(CT)中的人类微生物群进行分析和比较。此外,通过实时定量聚合酶链反应(qPCR)研究同一身体部位的丰度,以评估其与CRC的关联。观察到健康对照者与CRC患者之间细菌组成、丰度的差异以及与临床参数的关联。基于16S rRNA基因的分类分析显示存在三个主要细菌门,约占 reads 的80%:厚壁菌门(39.18%)、拟杆菌门(30.36%)和放线菌门(10.65%)。结果突出了不同细菌组成的存在;特别是,CRC患者的粪便样本似乎富含梭杆菌属,而在健康对照者的粪便样本中,拟杆菌门是检测到的主要门类之一,尽管这些差异无统计学意义。CT样本显示出最高的α多样性值。这些结果强调了CRC患者粪便与健康对照者相比在分类组成上的差异。尽管本研究纳入的样本数量较少,但这些结果表明微生物群在CRC进展中的重要性,并可能为CRC患者治疗干预措施的开发和新型微生物相关诊断工具铺平道路。