Proteomics Platform, Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
Department of Genome Sciences, University of Washington, Seattle, Seattle, Washington, USA.
Nat Methods. 2018 May;15(5):371-378. doi: 10.1038/nmeth.4643. Epub 2018 Apr 2.
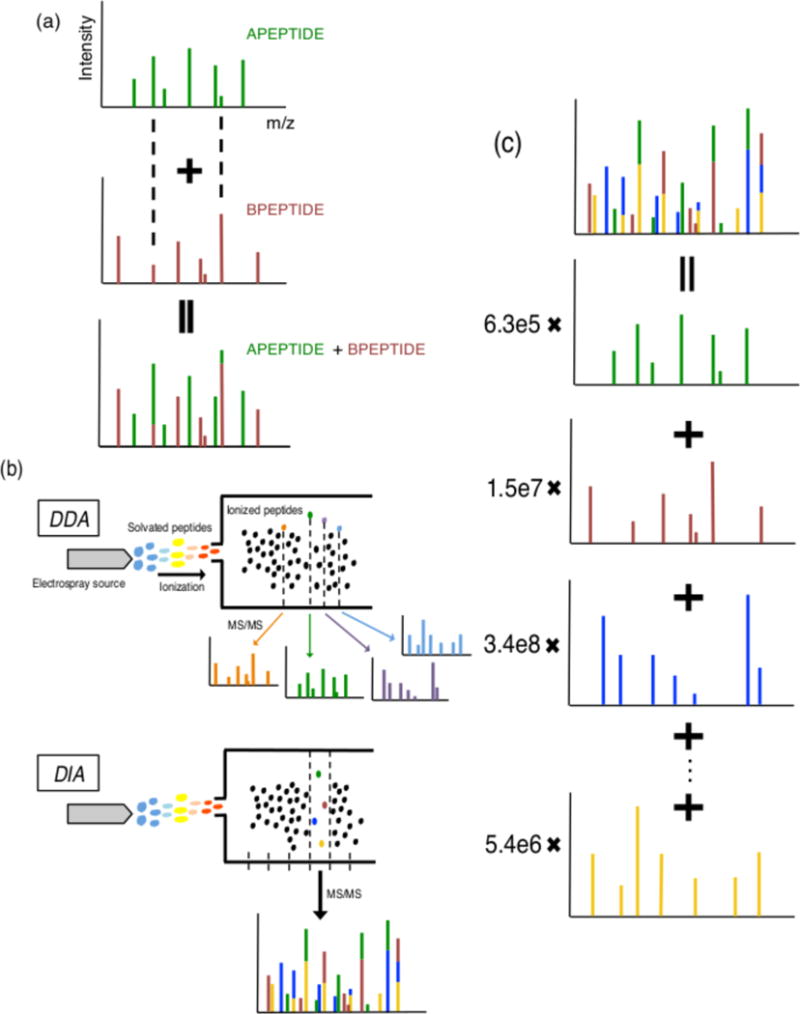
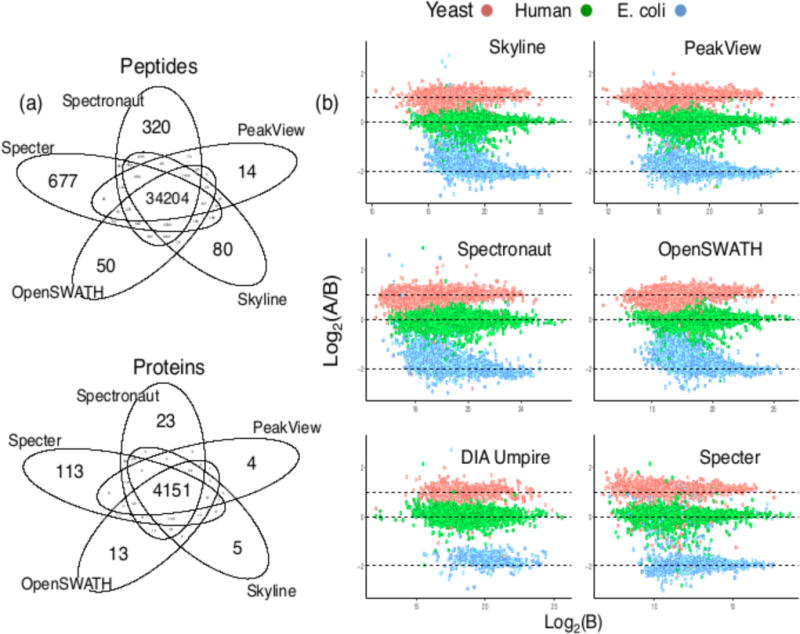
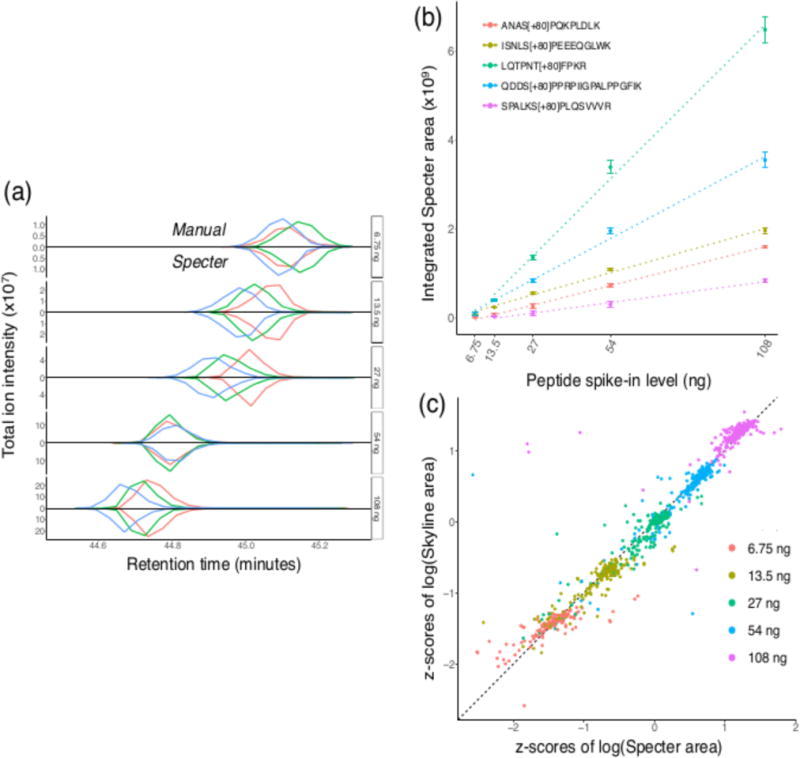
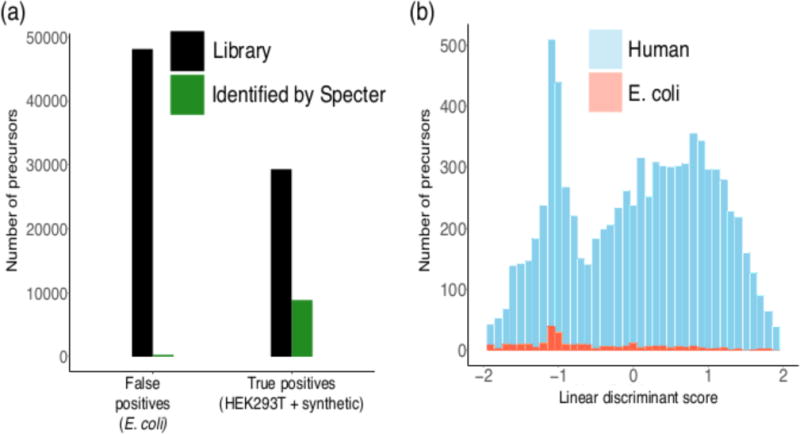
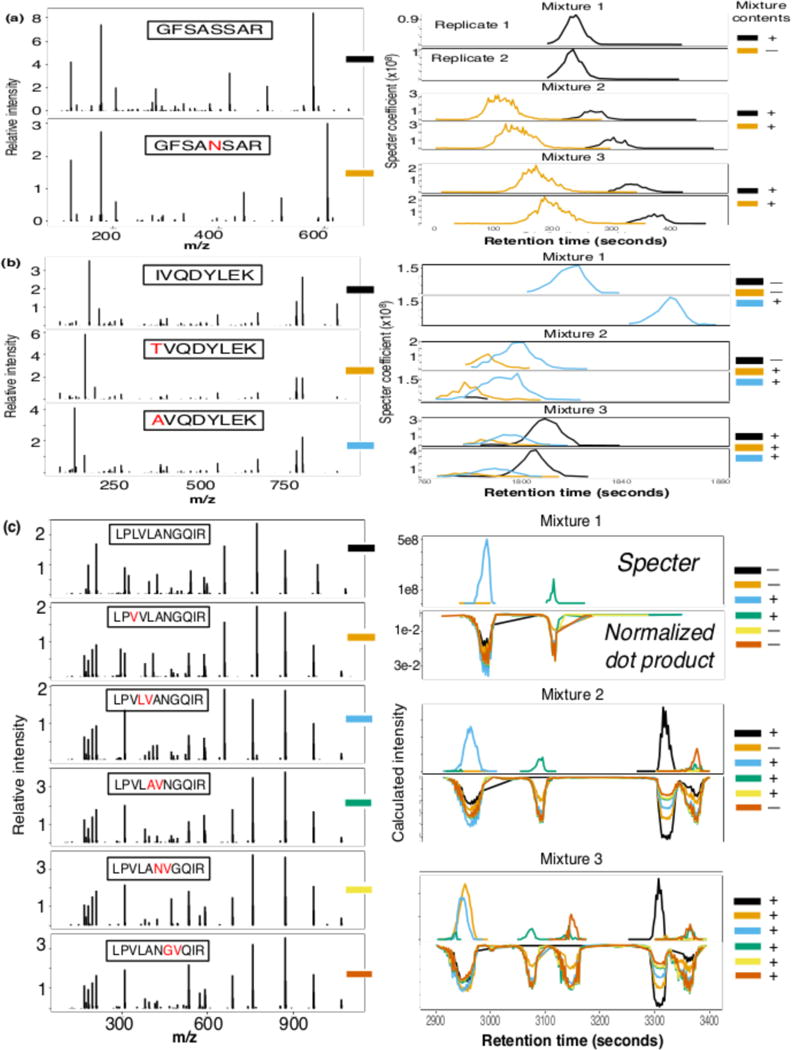
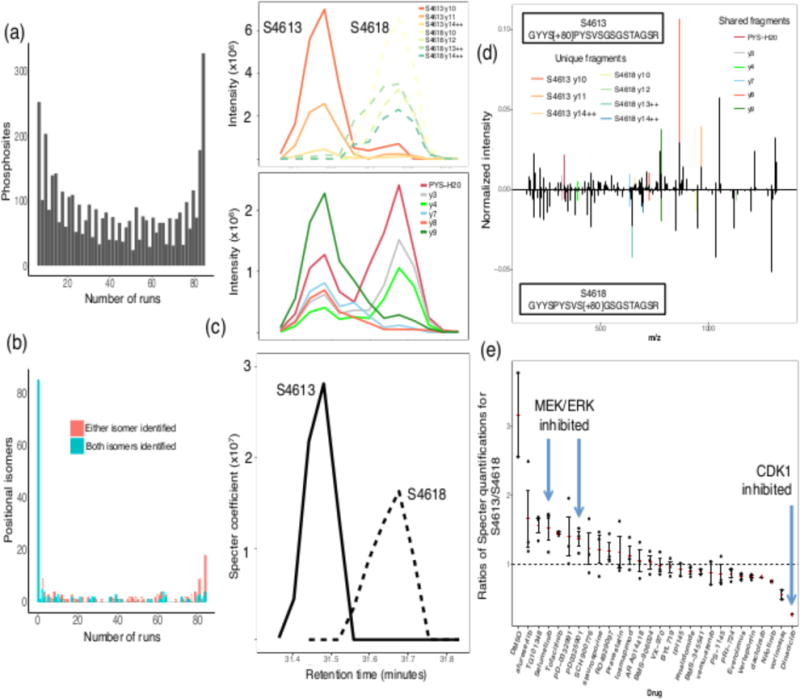
Mass spectrometry with data-independent acquisition (DIA) is a promising method to improve the comprehensiveness and reproducibility of targeted and discovery proteomics, in theory by systematically measuring all peptide precursors in a biological sample. However, the analytical challenges involved in discriminating between peptides with similar sequences in convoluted spectra have limited its applicability in important cases, such as the detection of single-nucleotide polymorphisms (SNPs) and alternative site localizations in phosphoproteomics data. We report Specter (https://github.com/rpeckner-broad/Specter), an open-source software tool that uses linear algebra to deconvolute DIA mixture spectra directly through comparison to a spectral library, thus circumventing the problems associated with typical fragment-correlation-based approaches. We validate the sensitivity of Specter and its performance relative to that of other methods, and show that Specter is able to successfully analyze cases involving highly similar peptides that are typically challenging for DIA analysis methods.
质谱分析与数据独立采集(DIA)是一种很有前途的方法,可以提高靶向和发现蛋白质组学的全面性和可重复性,理论上可以系统地测量生物样本中的所有肽前体。然而,在复杂的光谱中区分具有相似序列的肽的分析挑战限制了它在一些重要情况下的适用性,例如单核苷酸多态性(SNP)的检测和磷酸化蛋白质组学数据中替代位点的定位。我们报告了 Specter(https://github.com/rpeckner-broad/Specter),这是一种开源软件工具,它使用线性代数通过直接与光谱库进行比较来对 DIA 混合光谱进行去卷积,从而避免了与典型的基于片段相关性的方法相关的问题。我们验证了 Specter 的灵敏度及其相对于其他方法的性能,并表明 Specter 能够成功分析涉及高度相似肽的情况,这些情况通常对 DIA 分析方法具有挑战性。