Smith Robert W, van Rosmalen Rik P, Martins Dos Santos Vitor A P, Fleck Christian
Laboratory of Systems & Synthetic Biology, Wageningen UR, Stippeneng 4, Wageningen, 6708WE, The Netherlands.
LifeGlimmer GmbH, Markelstrasse 38, Berlin, 12163, Germany.
BMC Syst Biol. 2018 Jun 19;12(1):72. doi: 10.1186/s12918-018-0584-8.
Models of metabolism are often used in biotechnology and pharmaceutical research to identify drug targets or increase the direct production of valuable compounds. Due to the complexity of large metabolic systems, a number of conclusions have been drawn using mathematical methods with simplifying assumptions. For example, constraint-based models describe changes of internal concentrations that occur much quicker than alterations in cell physiology. Thus, metabolite concentrations and reaction fluxes are fixed to constant values. This greatly reduces the mathematical complexity, while providing a reasonably good description of the system in steady state. However, without a large number of constraints, many different flux sets can describe the optimal model and we obtain no information on how metabolite levels dynamically change. Thus, to accurately determine what is taking place within the cell, finer quality data and more detailed models need to be constructed.
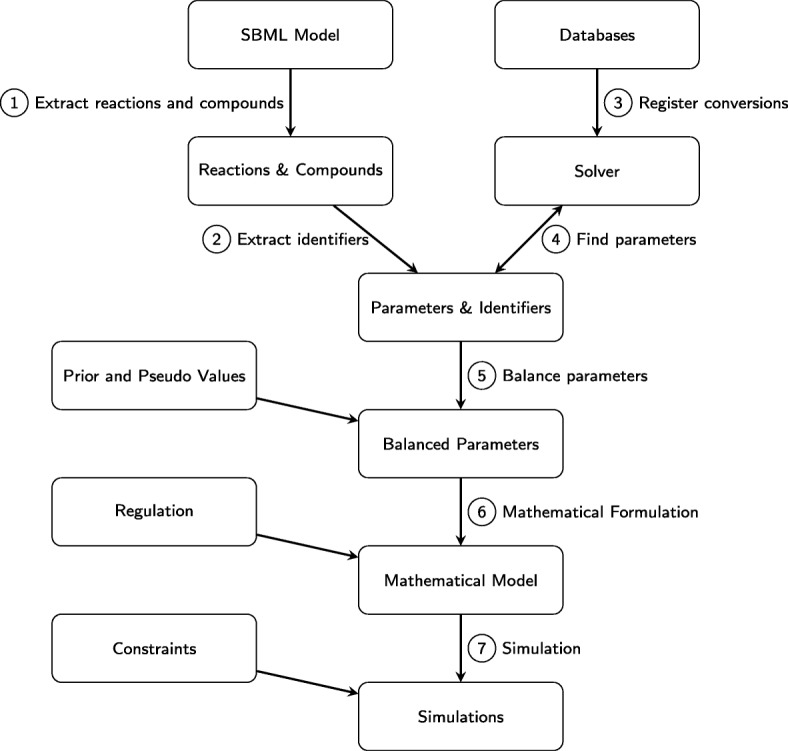
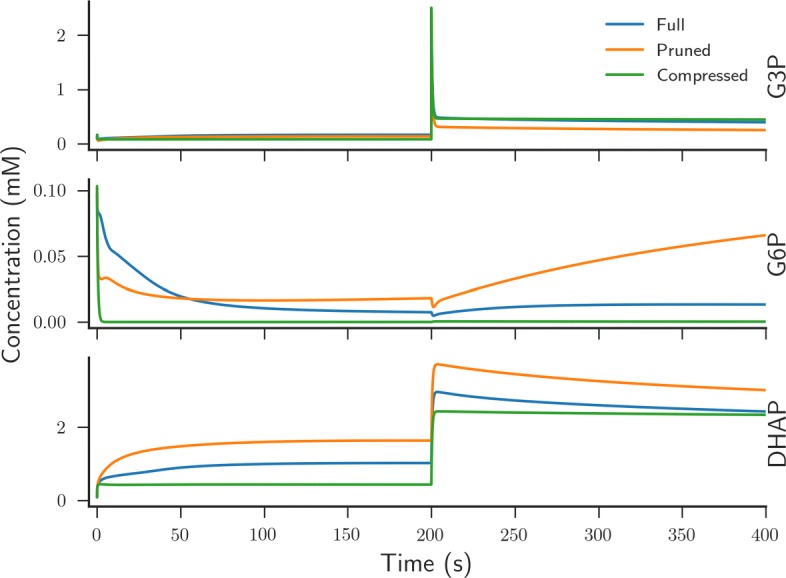
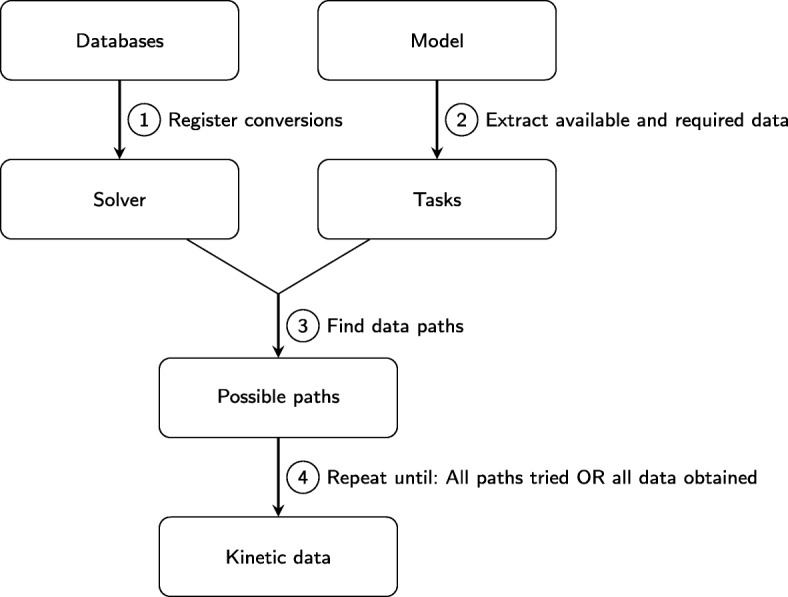
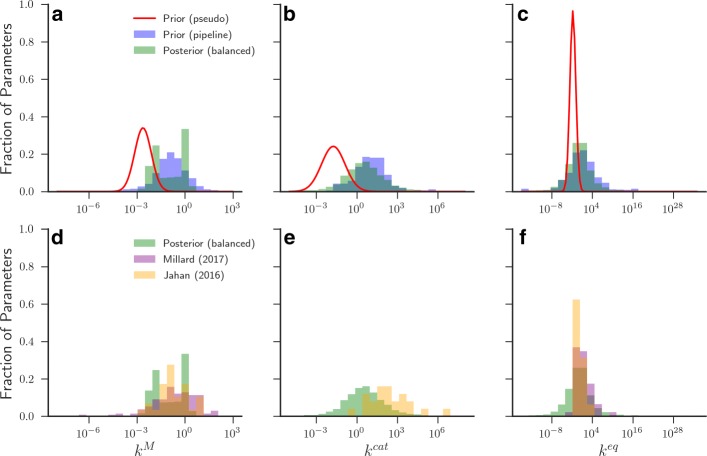
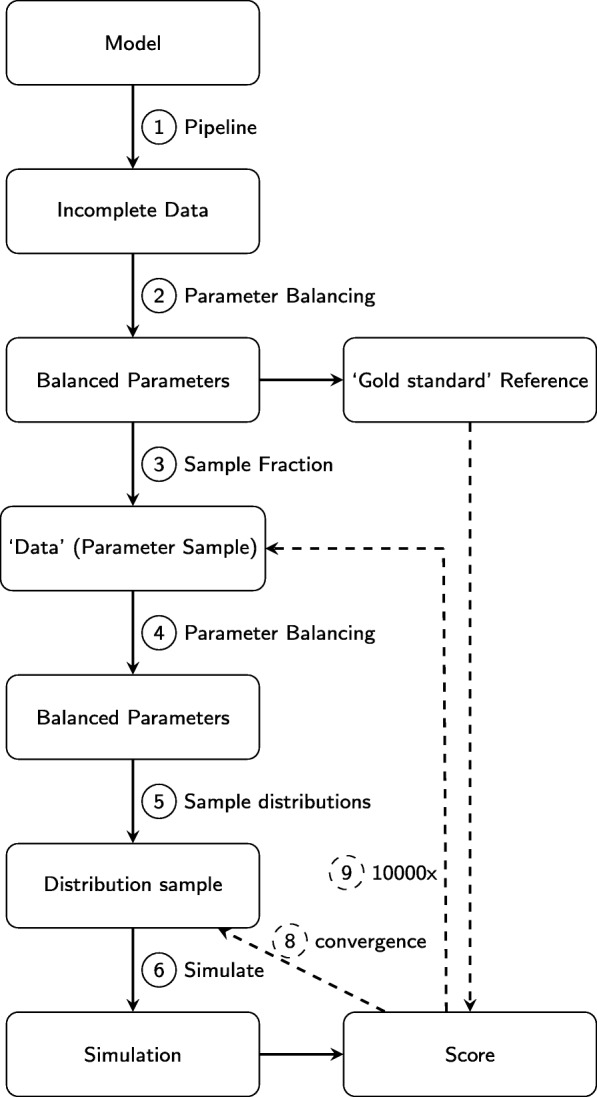
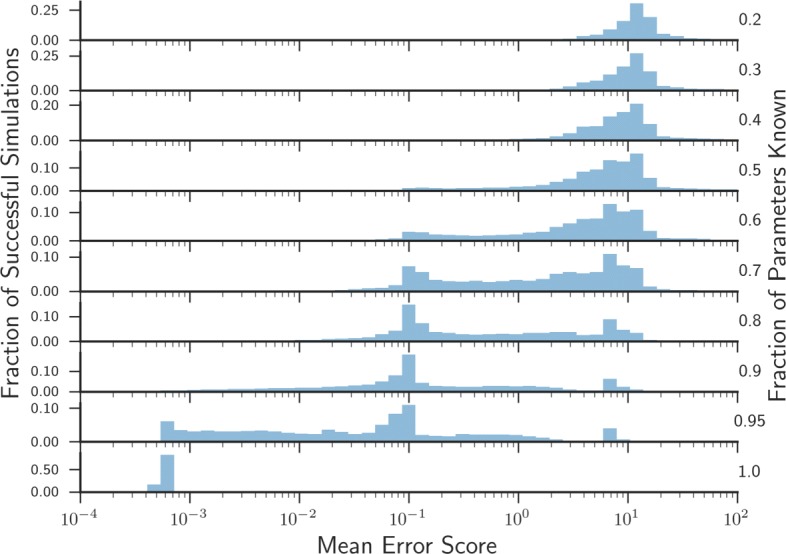
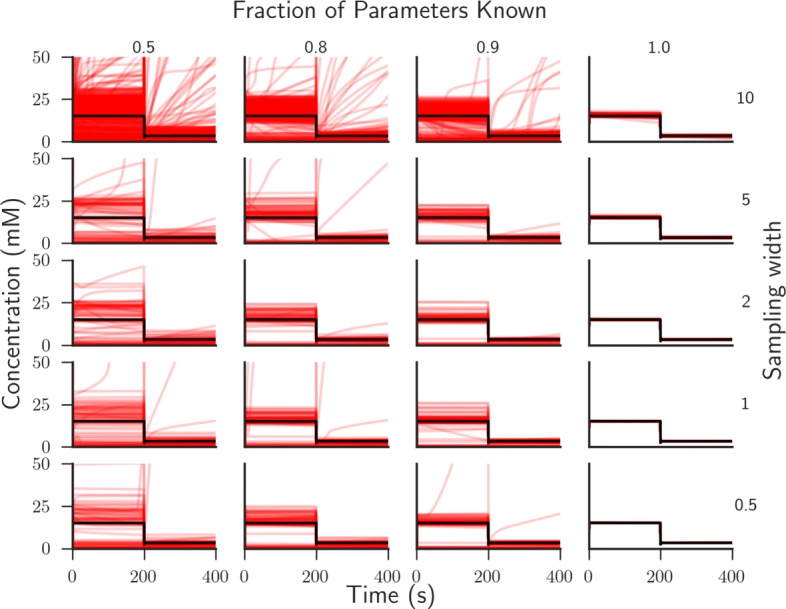
In this paper we present a computational framework, DMPy, that uses a network scheme as input to automatically search for kinetic rates and produce a mathematical model that describes temporal changes of metabolite fluxes. The parameter search utilises several online databases to find measured reaction parameters. From this, we take advantage of previous modelling efforts, such as Parameter Balancing, to produce an initial mathematical model of a metabolic pathway. We analyse the effect of parameter uncertainty on model dynamics and test how recent flux-based model reduction techniques alter system properties. To our knowledge this is the first time such analysis has been performed on large models of metabolism. Our results highlight that good estimates of at least 80% of the reaction rates are required to accurately model metabolic systems. Furthermore, reducing the size of the model by grouping reactions together based on fluxes alters the resulting system dynamics.
The presented pipeline automates the modelling process for large metabolic networks. From this, users can simulate their pathway of interest and obtain a better understanding of how altering conditions influences cellular dynamics. By testing the effects of different parameterisations we are also able to provide suggestions to help construct more accurate models of complete metabolic systems in the future.
代谢模型常用于生物技术和药物研究,以识别药物靶点或提高有价值化合物的直接产量。由于大型代谢系统的复杂性,人们使用了一些带有简化假设的数学方法得出了许多结论。例如,基于约束的模型描述的内部浓度变化比细胞生理变化快得多。因此,代谢物浓度和反应通量被固定为恒定值。这大大降低了数学复杂性,同时在稳态下对系统提供了相当好的描述。然而,没有大量的约束条件,许多不同的通量集可以描述最优模型,并且我们无法获得代谢物水平如何动态变化的信息。因此,为了准确确定细胞内发生的情况,需要构建质量更高的数据和更详细的模型。
在本文中,我们提出了一个计算框架DMPy,它使用网络方案作为输入,自动搜索动力学速率并生成一个描述代谢物通量随时间变化的数学模型。参数搜索利用几个在线数据库来查找测量的反应参数。据此,我们利用先前的建模成果,如参数平衡,来生成代谢途径的初始数学模型。我们分析了参数不确定性对模型动力学的影响,并测试了最近基于通量的模型简化技术如何改变系统特性。据我们所知,这是首次对大型代谢模型进行此类分析。我们的结果表明,要准确模拟代谢系统,至少需要80%的反应速率的良好估计值。此外,基于通量将反应分组来减小模型规模会改变所得系统的动力学。
所提出的流程自动化了大型代谢网络的建模过程。据此,用户可以模拟他们感兴趣的途径,并更好地理解改变条件如何影响细胞动力学。通过测试不同参数化的效果,我们还能够提供建议,以帮助未来构建更准确的完整代谢系统模型。