A Duarte Matheus, F Silva João M, R Brito Clara, S Teixeira Danilo, L Melo Fernando, M Ribeiro Bergmann, Nagata Tatsuya, S Campos Fabrício
Faculdade de Agronomia e Veterinária, Universidade de Brasília, Brasília-DF 70.910-900, Brazil.
Departamento de Biologia Celular, Instituto de Biologia, Universidade de Brasília, Brasília-DF 70.910-900, Brazil.
Viruses. 2019 Aug 30;11(9):803. doi: 10.3390/v11090803.
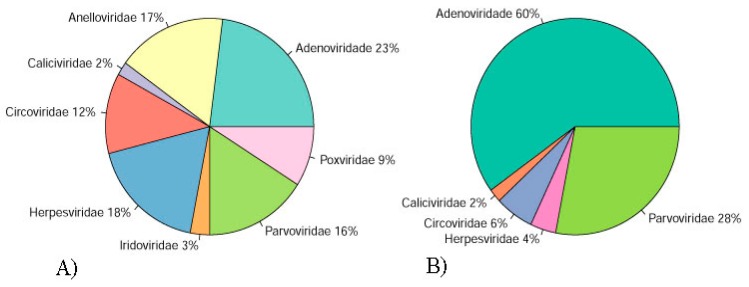
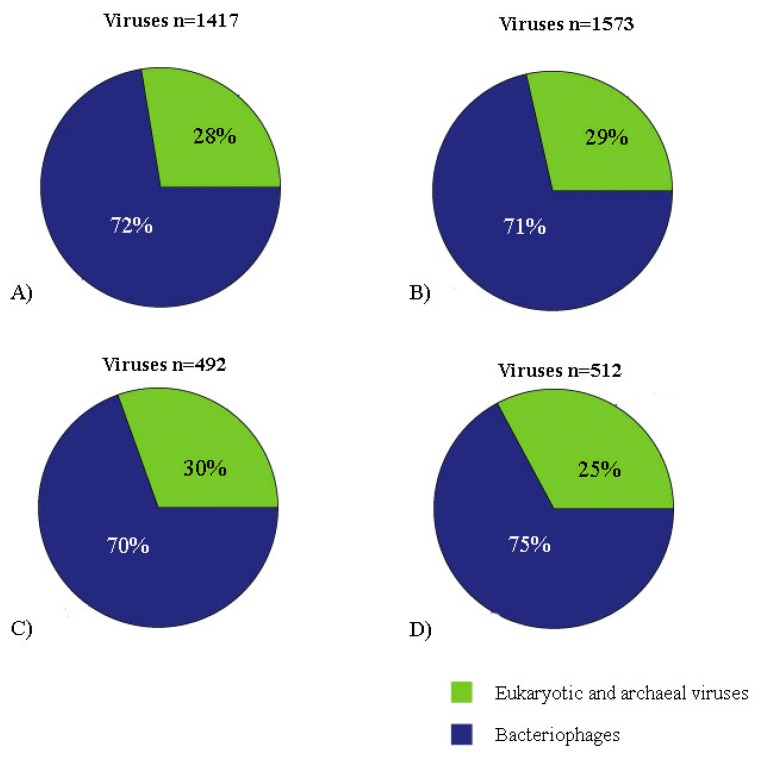
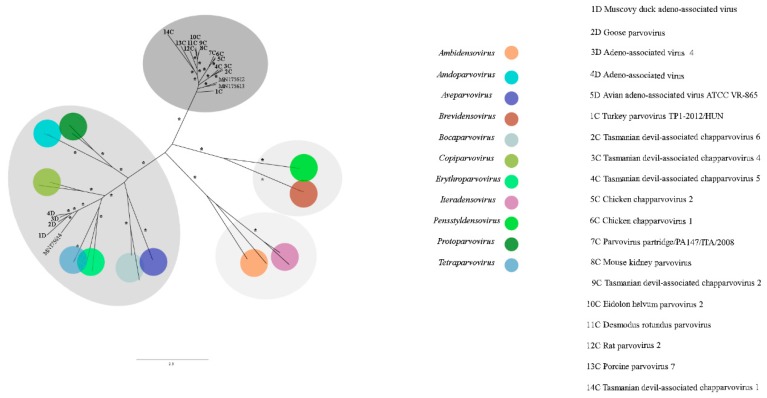
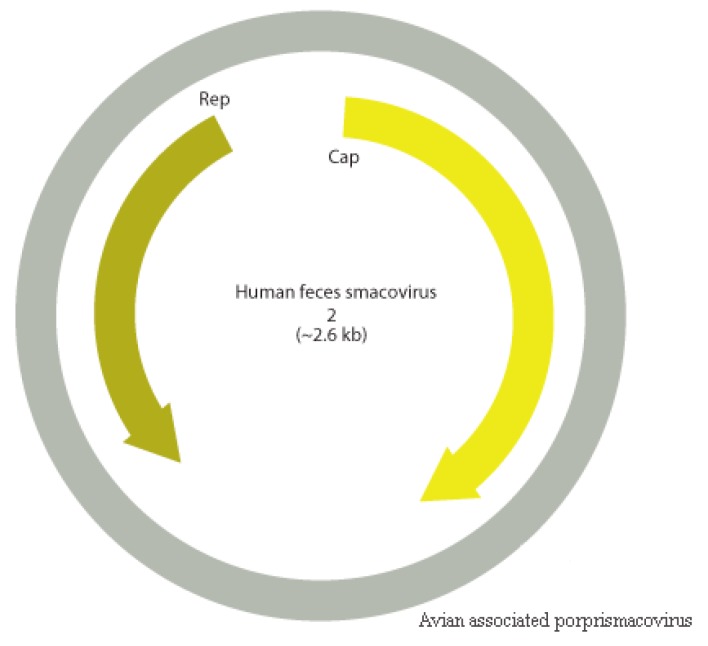
The Brazilian Cerrado fauna shows very wide diversity and can be a potential viral reservoir. Therefore, the animal's susceptibility to some virus can serve as early warning signs of potential human virus diseases. Moreover, the wild animal virome of this biome is unknown. Based on this scenario, high-throughput sequencing contributes a robust tool for the identification of known and unknown virus species in this environment. In the present study, faeces samples from cerrado birds (, , and ) and mammals (, , and ) were collected at the Veterinary Hospital, University of Brasília. Viral nucleic acid was extracted, submitted to random amplification, and sequenced by Illumina HiSeq platform. The reads were de novo assembled, and the identities of the contigs were evaluated by Blastn and tblastx searches. Most viral contigs analyzed were closely related to bacteriophages. Novel archaeal viruses of the family were detected. Moreover, sequences of members of , , , , and families were identified. Complete and nearly complete genomes of known anelloviruses, circoviruses, and parvoviruses were obtained, as well as putative novel species. We demonstrate that the metagenomics approach applied in this work was effective for identification of known and putative new viruses in faeces samples from Brazilian Cerrado fauna.
巴西塞拉多地区的动物群表现出非常广泛的多样性,可能是一个潜在的病毒库。因此,动物对某些病毒的易感性可作为潜在人类病毒疾病的早期预警信号。此外,这个生物群落的野生动物病毒组尚不清楚。基于这种情况,高通量测序为识别该环境中已知和未知病毒物种提供了一个强大的工具。在本研究中,从巴西利亚大学兽医院采集了塞拉多地区鸟类(、和)和哺乳动物(、和)的粪便样本。提取病毒核酸,进行随机扩增,并通过Illumina HiSeq平台进行测序。对 reads 进行从头组装,并通过Blastn和tblastx搜索评估重叠群的身份。分析的大多数病毒重叠群与噬菌体密切相关。检测到了科的新型古病毒。此外,还鉴定了、、、和科成员的序列。获得了已知环病毒、圆环病毒和细小病毒的完整和近乎完整的基因组,以及推定的新物种。我们证明,本研究中应用的宏基因组学方法对于识别巴西塞拉多地区动物粪便样本中的已知和推定新病毒是有效的。