Kouchaki Samaneh, Yang Yang, Lachapelle Alexander, Walker Timothy M, Walker A Sarah, Peto Timothy E A, Crook Derrick W, Clifton David A
Department of Engineering Science, Institute of Biomedical Engineering, University of Oxford, Oxford, United Kingdom.
Oxford-Suzhou Centre for Advanced Research, Suzhou, China.
Front Microbiol. 2020 Apr 22;11:667. doi: 10.3389/fmicb.2020.00667. eCollection 2020.
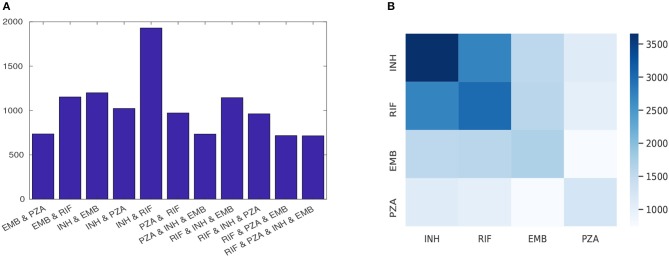
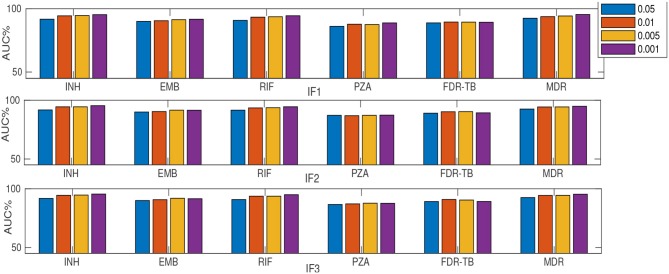
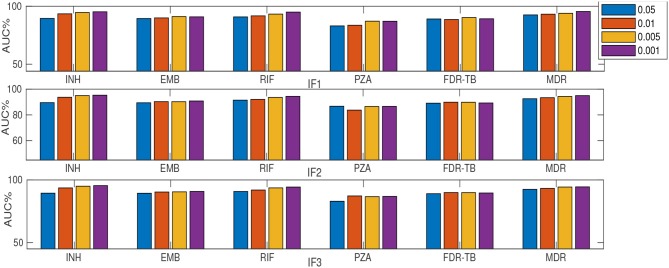
Resistance prediction and mutation ranking are important tasks in the analysis of Tuberculosis sequence data. Due to standard regimens for the use of first-line antibiotics, resistance co-occurrence, in which samples are resistant to multiple drugs, is common. Analysing all drugs simultaneously should therefore enable patterns reflecting resistance co-occurrence to be exploited for resistance prediction. Here, multi-label random forest (MLRF) models are compared with single-label random forest (SLRF) for both predicting phenotypic resistance from whole genome sequences and identifying important mutations for better prediction of four first-line drugs in a dataset of 13402 isolates. Results confirmed that MLRFs can improve performance compared to conventional clinical methods (by 18.10%) and SLRFs (by 0.91%). In addition, we identified a list of candidate mutations that are important for resistance prediction or that are related to resistance co-occurrence. Moreover, we found that retraining our analysis to a subset of top-ranked mutations was sufficient to achieve satisfactory performance. The source code can be found at http://www.robots.ox.ac.uk/~davidc/code.php.
耐药性预测和突变排序是结核病序列数据分析中的重要任务。由于一线抗生素的标准使用方案,样本对多种药物耐药的耐药共现情况很常见。因此,同时分析所有药物应能利用反映耐药共现的模式进行耐药性预测。在此,在一个包含13402个分离株的数据集里,将多标签随机森林(MLRF)模型与单标签随机森林(SLRF)进行比较,以从全基因组序列预测表型耐药性,并识别重要突变以更好地预测四种一线药物。结果证实,与传统临床方法相比,MLRF可提高性能(提高18.10%),与SLRF相比也可提高性能(提高0.91%)。此外,我们确定了一系列对耐药性预测很重要或与耐药共现相关的候选突变。而且,我们发现将分析重新训练到排名靠前的突变子集足以获得令人满意的性能。源代码可在http://www.robots.ox.ac.uk/~davidc/code.php找到。