Department of Genetics, Harvard Medical School, Boston, MA, USA.
Department of Human Evolutionary Biology, Harvard University, Cambridge, MA, USA.
Mol Ecol Resour. 2020 Nov;20(6):1658-1667. doi: 10.1111/1755-0998.13230. Epub 2020 Aug 19.
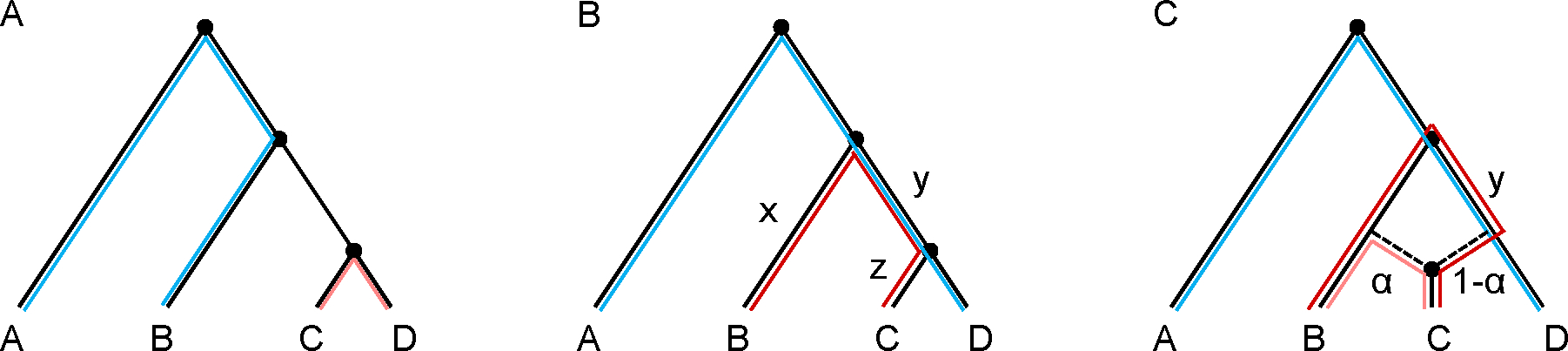
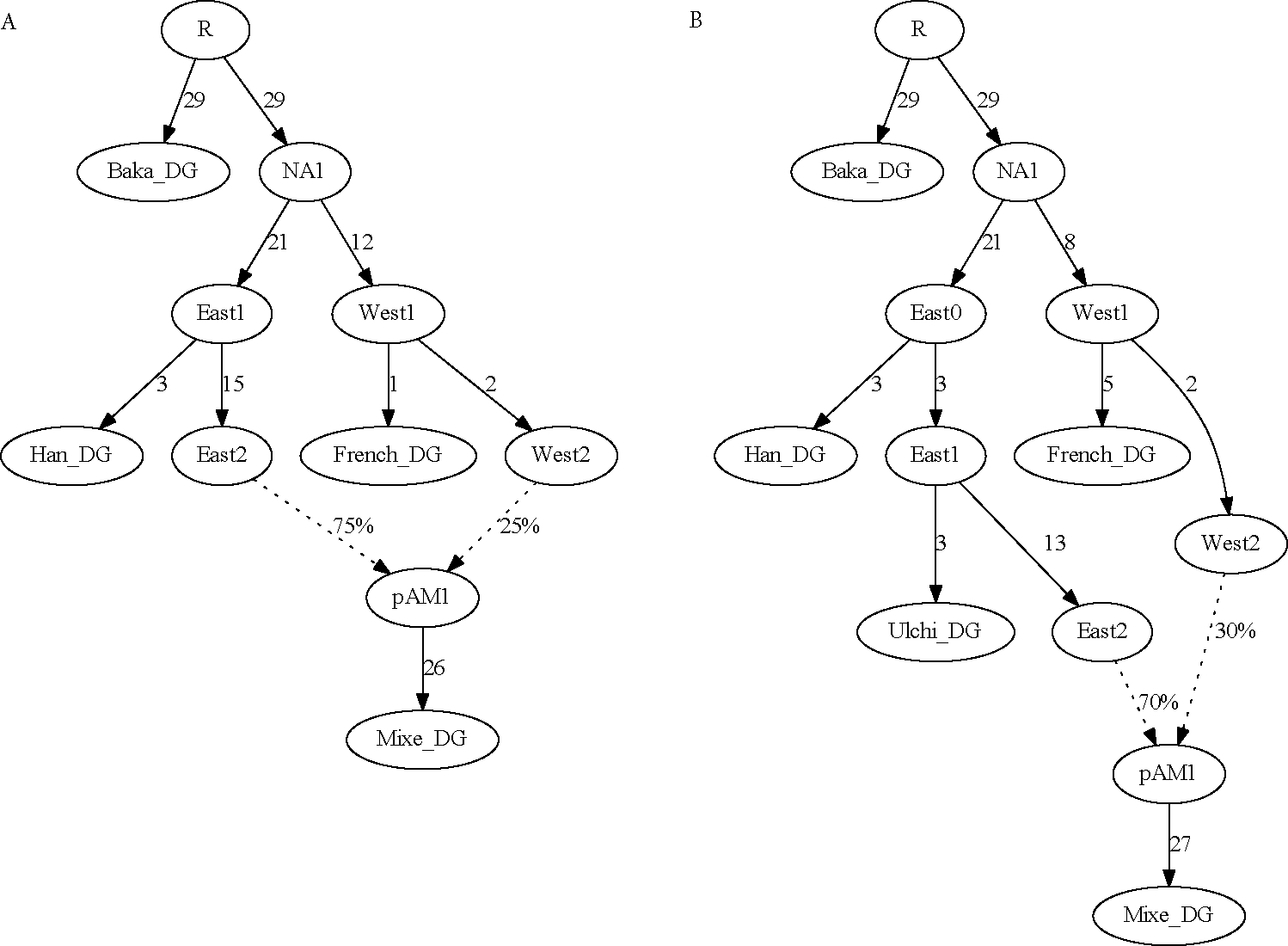
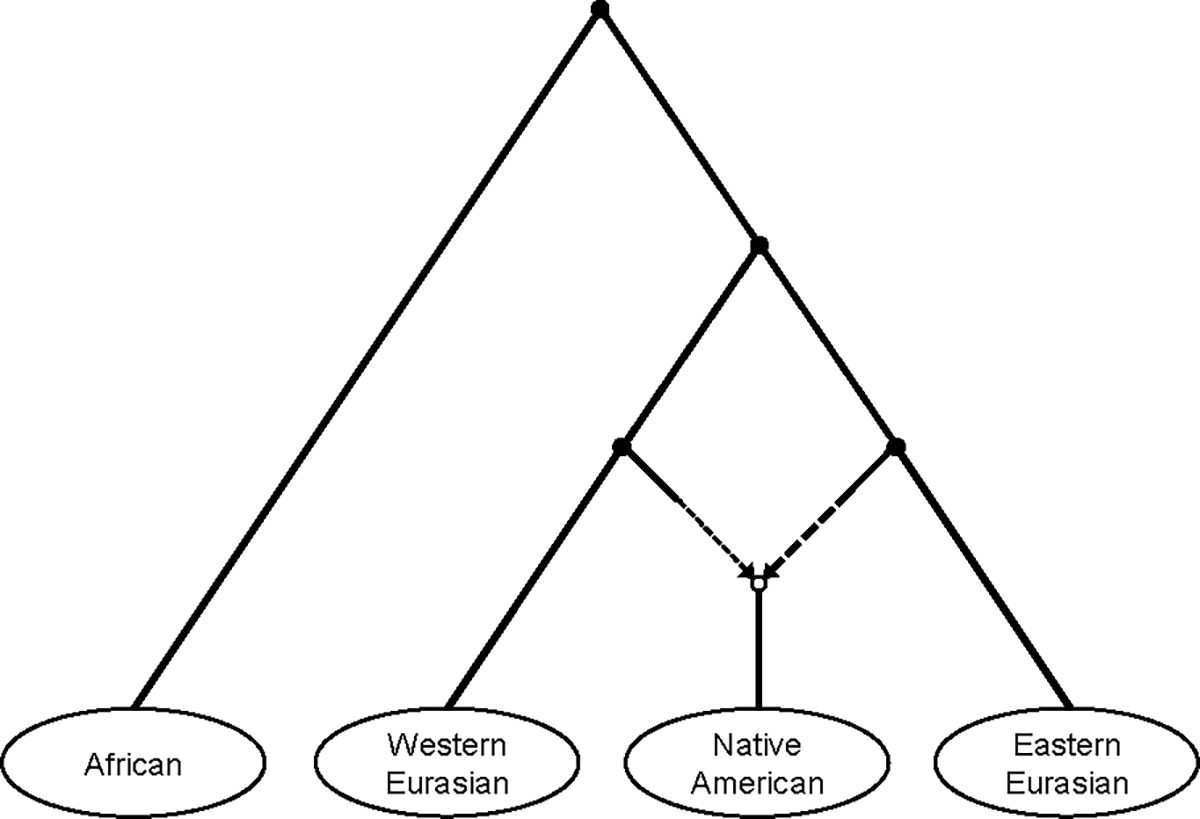
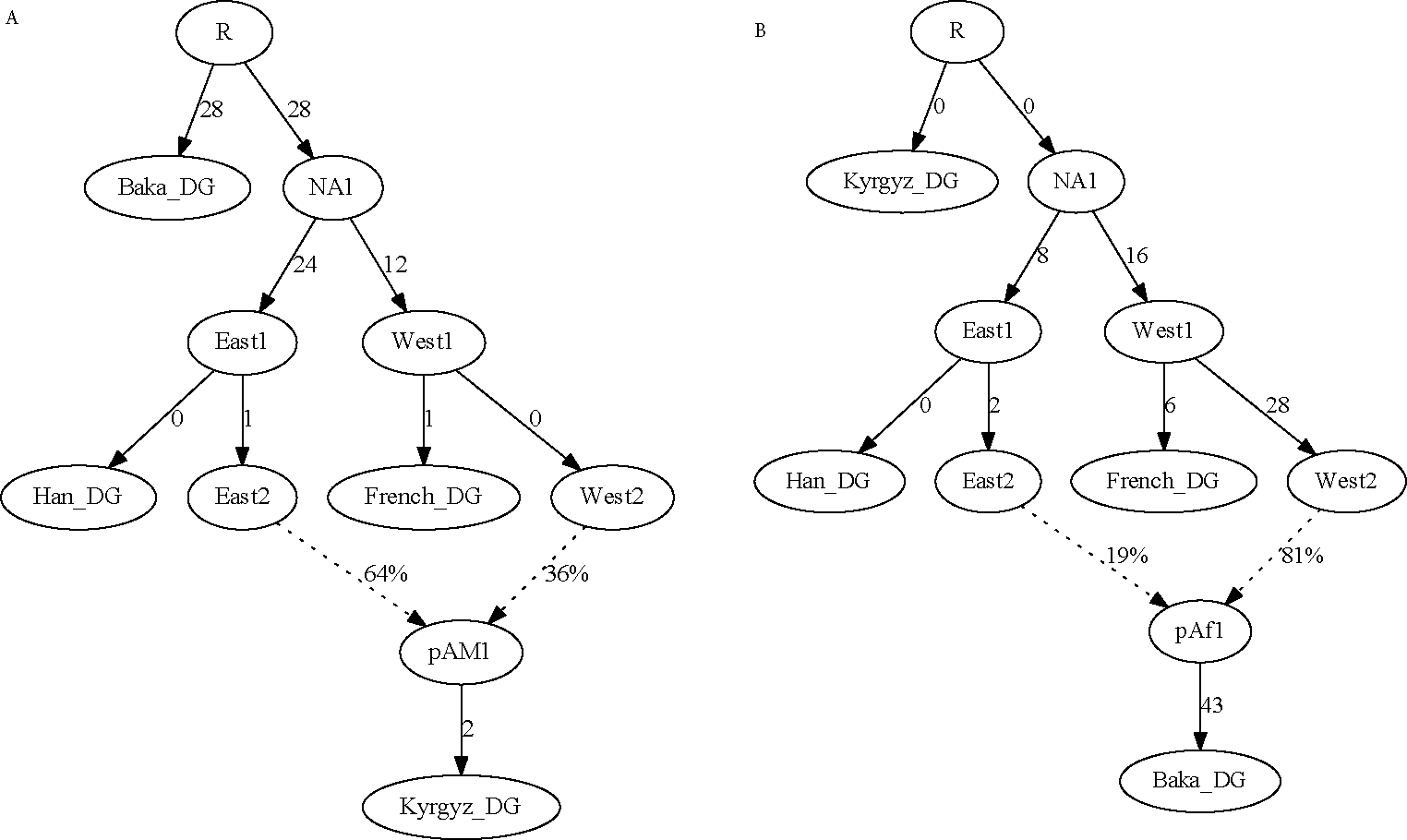
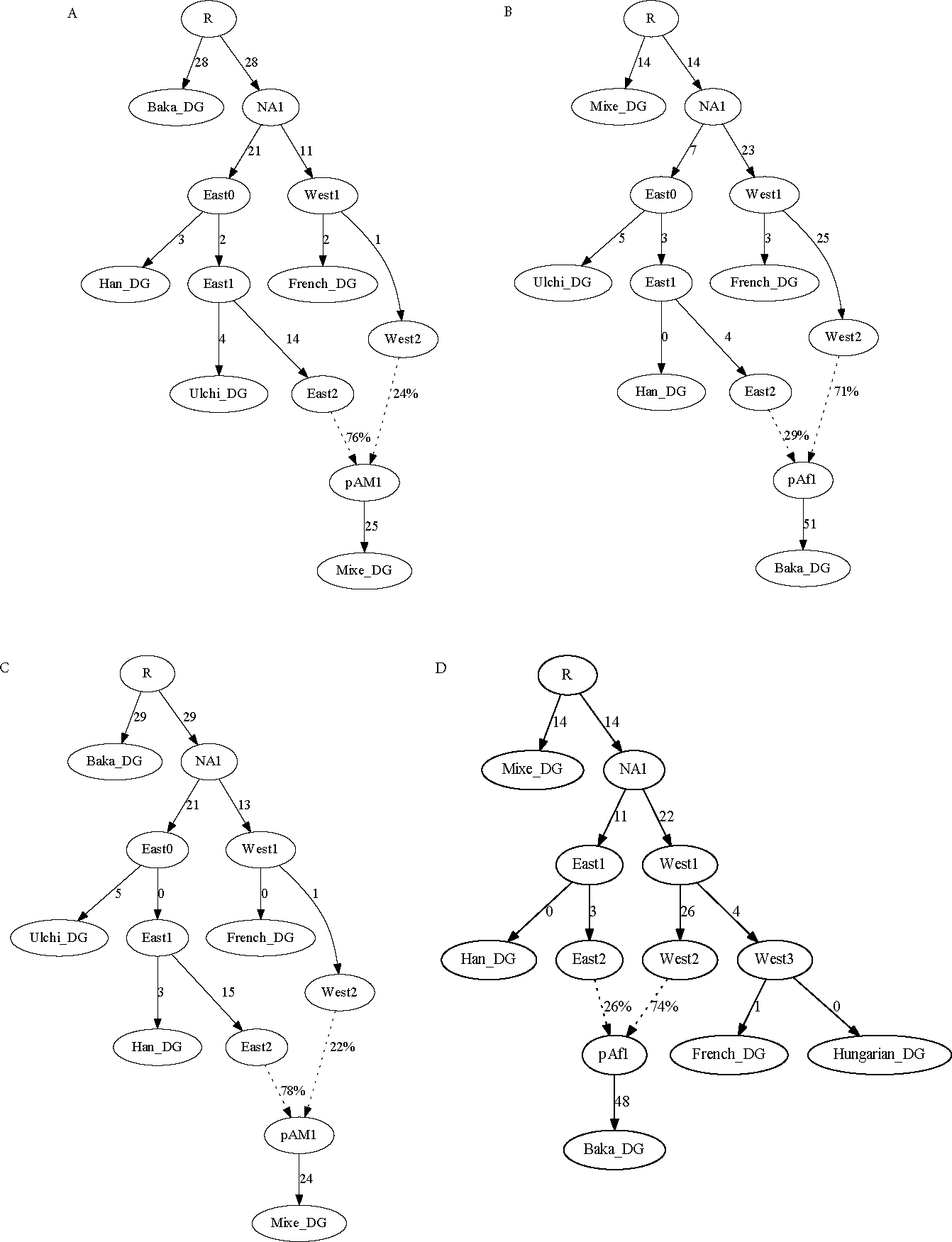
A popular approach to learning about admixture from population genetic data is by computing the allele-sharing summary statistics known as f-statistics. Compared to some methods in population genetics, f-statistics are relatively simple, but interpreting them can still be complicated at times. In addition, f-statistics can be used to build admixture graphs (multi-population trees allowing for admixture events), which provide more explicit and thorough modelling capabilities but are correspondingly more complex to work with. Here, I discuss some of these issues to provide users of these tools with a basic guide for protocols and procedures. My focus is on the kinds of conclusions that can or cannot be drawn from the results of f -statistics and admixture graphs, illustrated with real-world examples involving human populations.
从群体遗传学数据中了解混合的一种流行方法是计算等位基因共享汇总统计量,即 f 统计量。与群体遗传学中的一些方法相比,f 统计量相对简单,但有时解释它们仍然很复杂。此外,f 统计量可用于构建混合图(允许混合事件的多群体树),这提供了更明确和彻底的建模能力,但相应地更复杂。在这里,我将讨论其中的一些问题,为这些工具的用户提供协议和程序的基本指南。我的重点是从 f 统计量和混合图的结果中可以或不能得出什么样的结论,并结合涉及人类群体的实际例子来说明。