Gillis Sierra, Roth Andrew
Department of Molecular Oncology, BC Cancer Research Institute, 675 W 10th Ave, Vancouver, V5Z 1L3, Canada.
Department of Computer Science, University of British Columbia, 2366 Main Mall, Vancouver, V6T 1Z4, Canada.
BMC Bioinformatics. 2020 Dec 10;21(1):571. doi: 10.1186/s12859-020-03919-2.
At diagnosis tumours are typically composed of a mixture of genomically distinct malignant cell populations. Bulk sequencing of tumour samples coupled with computational deconvolution can be used to identify these populations and study cancer evolution. Existing computational methods for populations deconvolution are slow and/or potentially inaccurate when applied to large datasets generated by whole genome sequencing data.
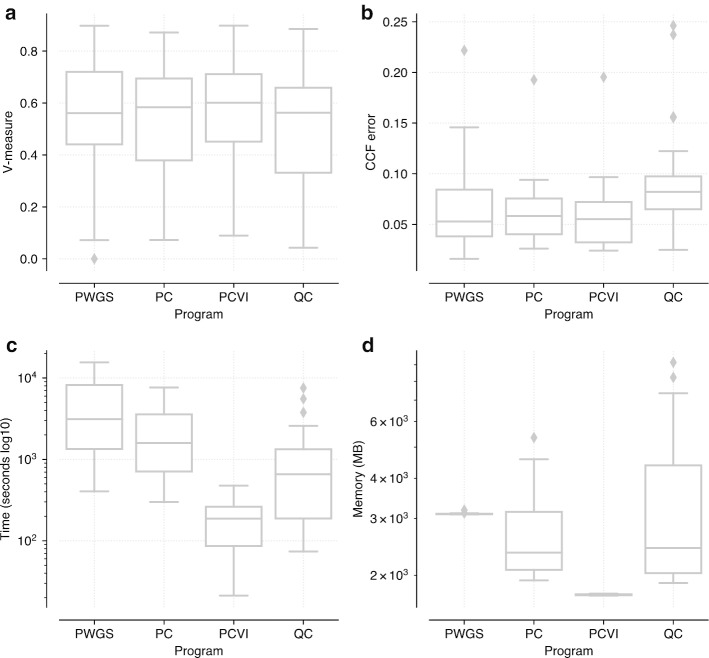
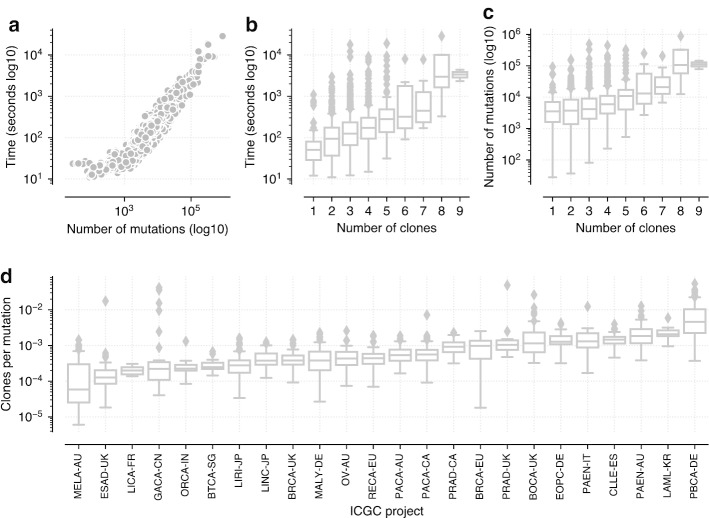
We describe PyClone-VI, a computationally efficient Bayesian statistical method for inferring the clonal population structure of cancers. We demonstrate the utility of the method by analyzing data from 1717 patients from PCAWG study and 100 patients from the TRACERx study.
Our proposed method is 10-100× times faster than existing methods, while providing results which are as accurate. Software implementing our method is freely available https://github.com/Roth-Lab/pyclone-vi .
在肿瘤诊断时,肿瘤通常由基因组不同的恶性细胞群体混合组成。肿瘤样本的批量测序结合计算反卷积可用于识别这些群体并研究癌症进化。现有的用于群体反卷积的计算方法在应用于由全基因组测序数据生成的大型数据集时速度慢且/或可能不准确。
我们描述了PyClone-VI,一种用于推断癌症克隆群体结构的计算高效的贝叶斯统计方法。我们通过分析来自PCAWG研究的1717名患者和TRACERx研究的100名患者的数据来证明该方法的实用性。
我们提出的方法比现有方法快10-100倍,同时提供同样准确的结果。实现我们方法的软件可在https://github.com/Roth-Lab/pyclone-vi上免费获取。