Department of Chemistry, University of California, Irvine, California 92697-2025, United States.
Acc Chem Res. 2021 Mar 16;54(6):1347-1359. doi: 10.1021/acs.accounts.0c00809. Epub 2021 Feb 17.
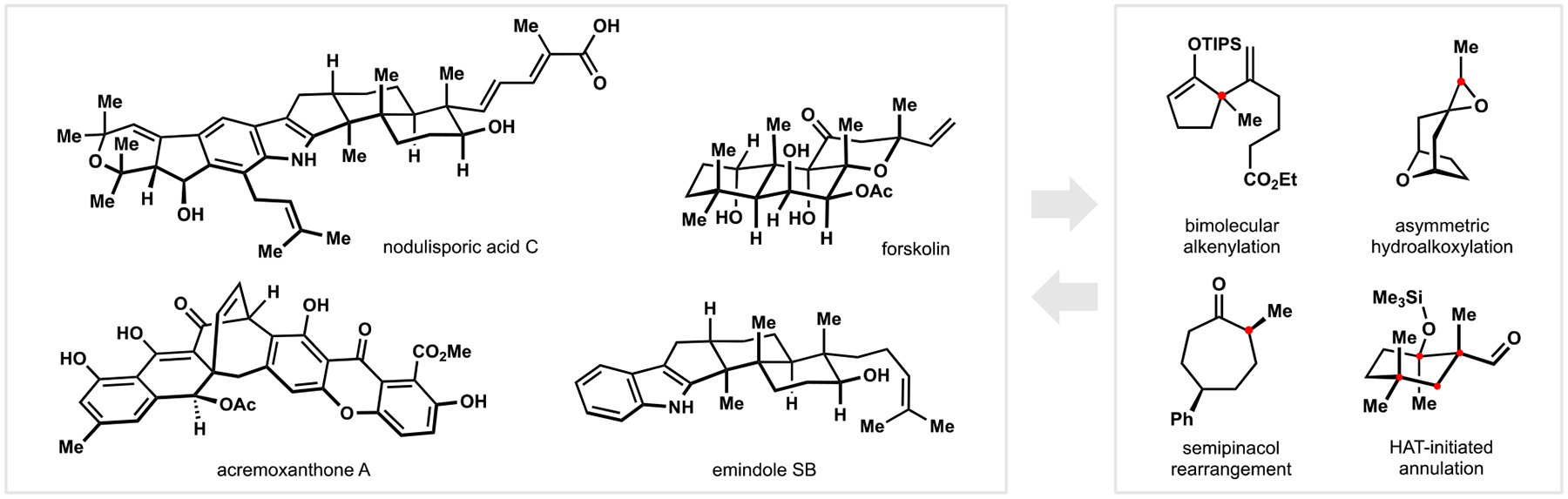
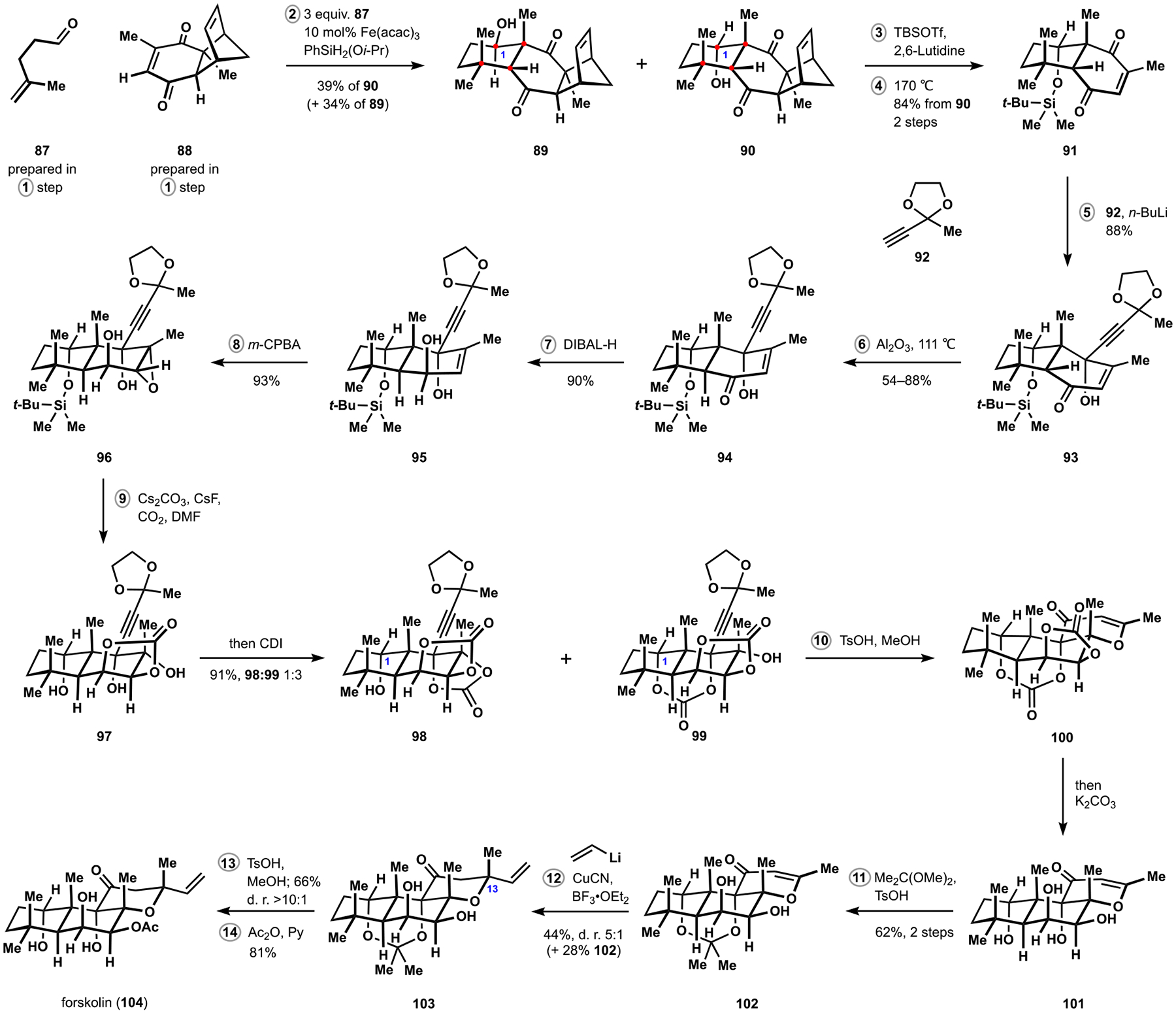
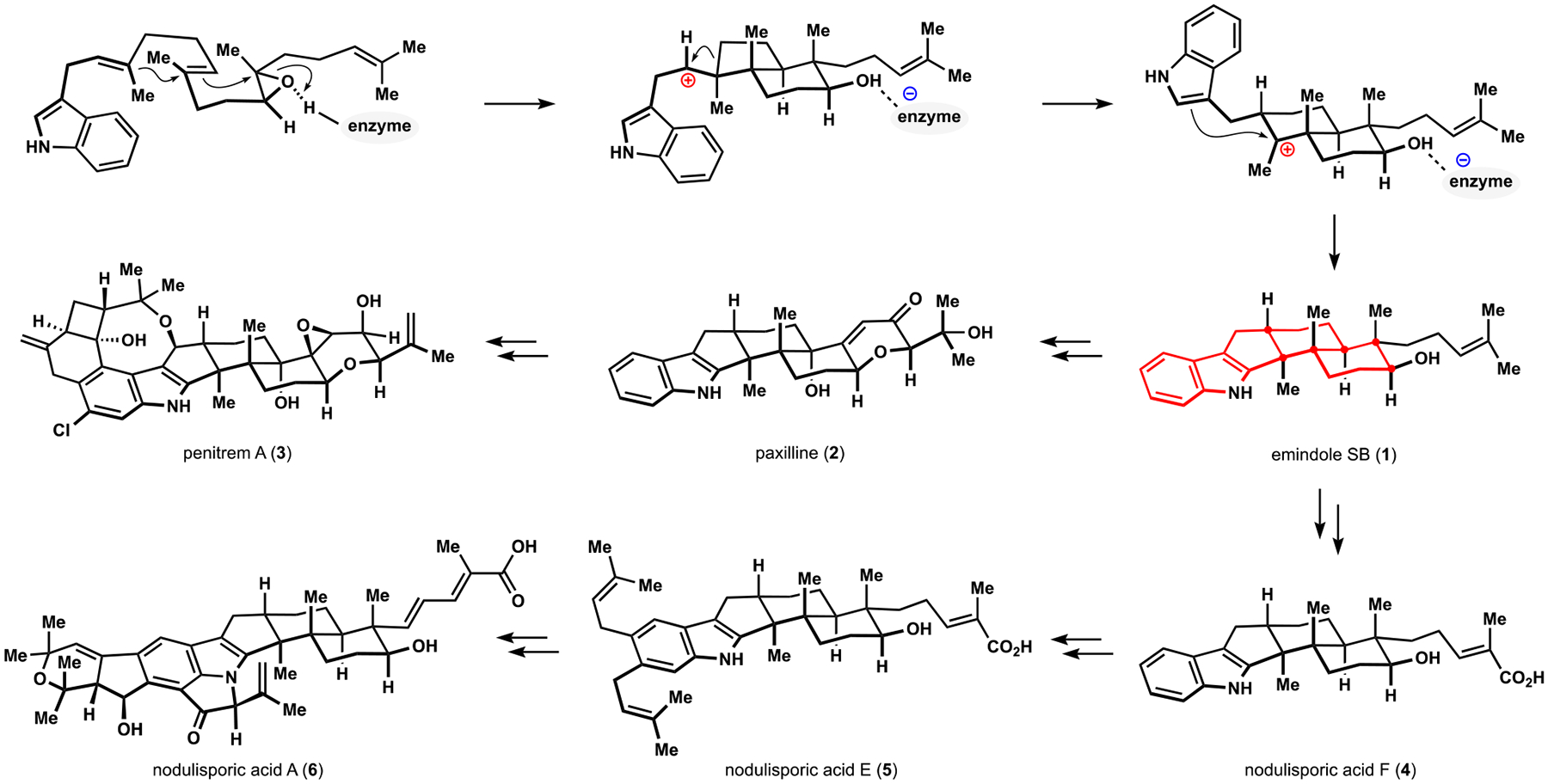
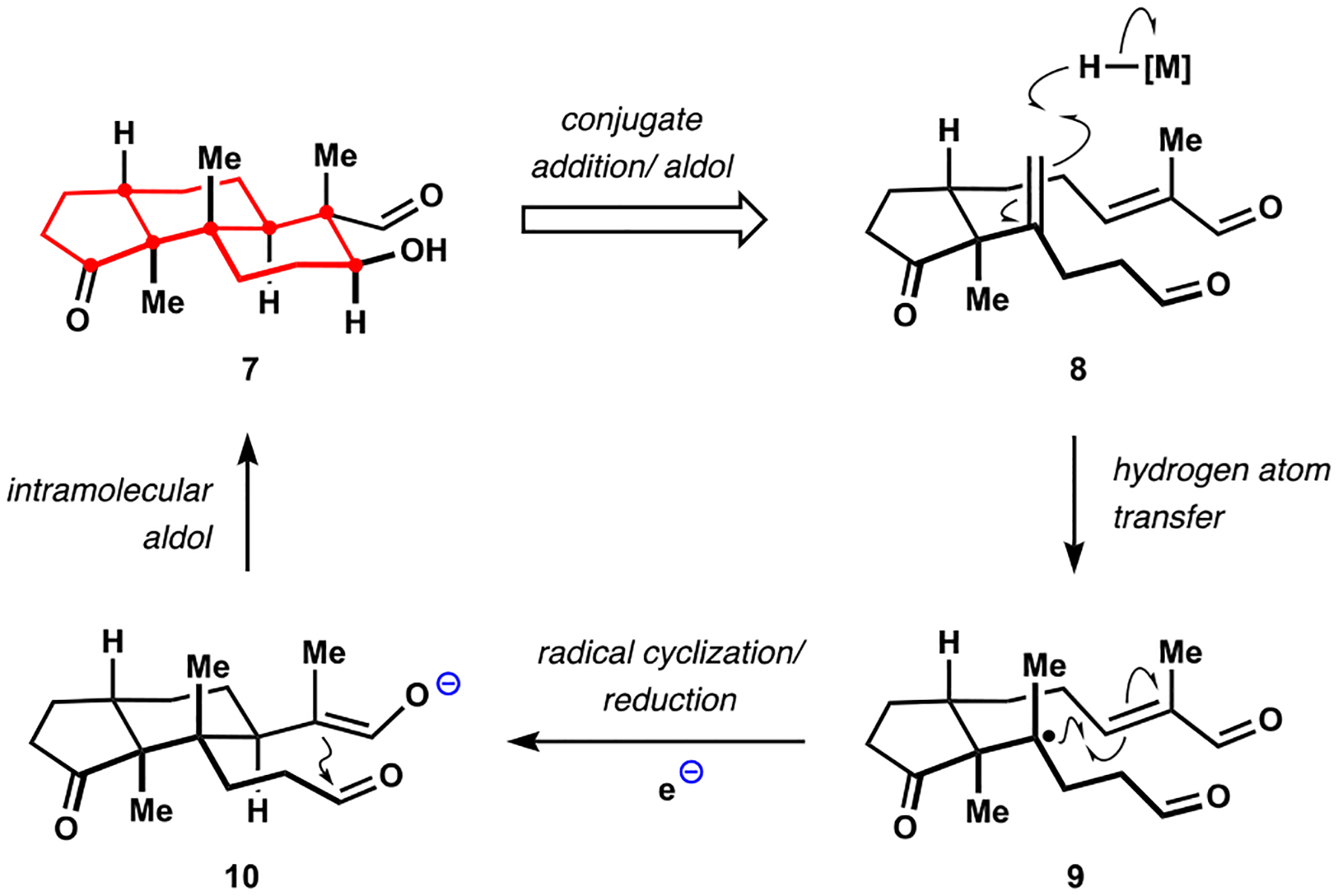
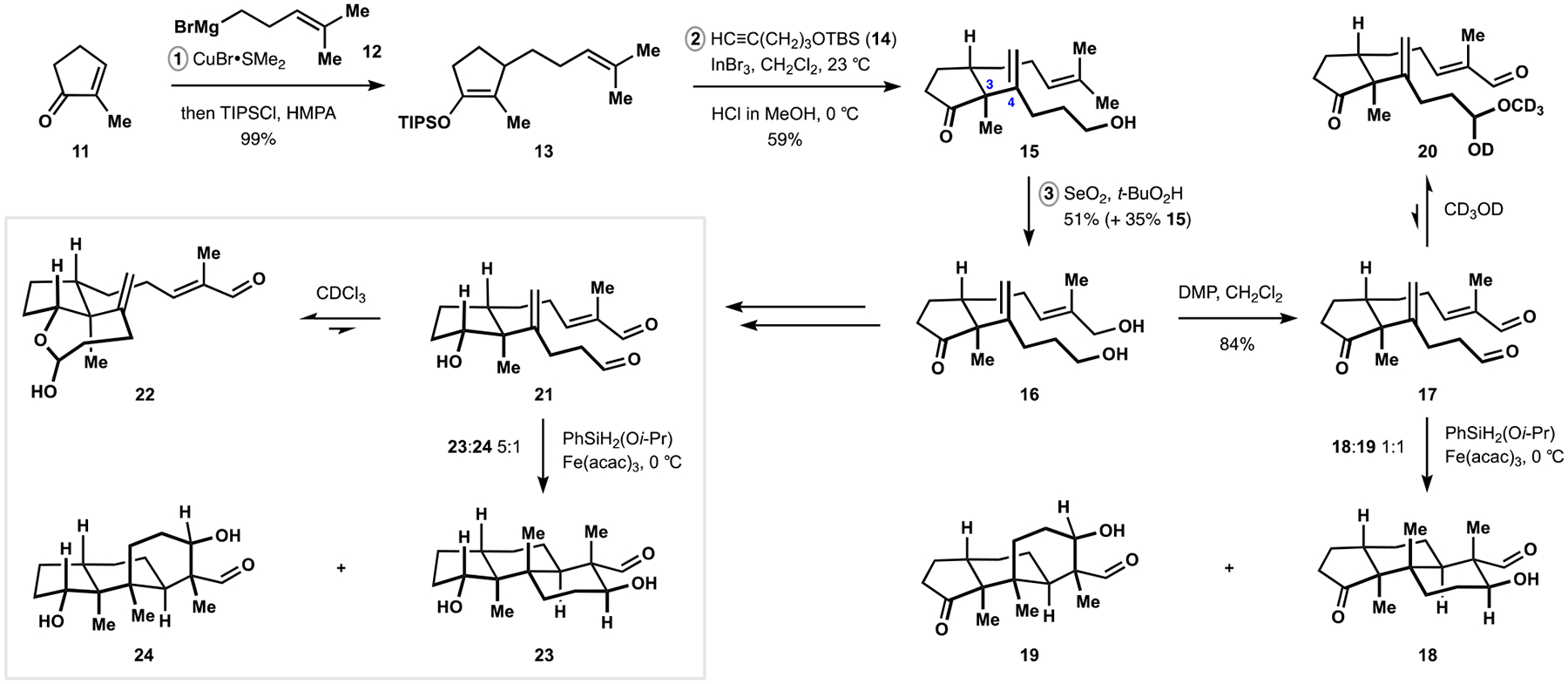
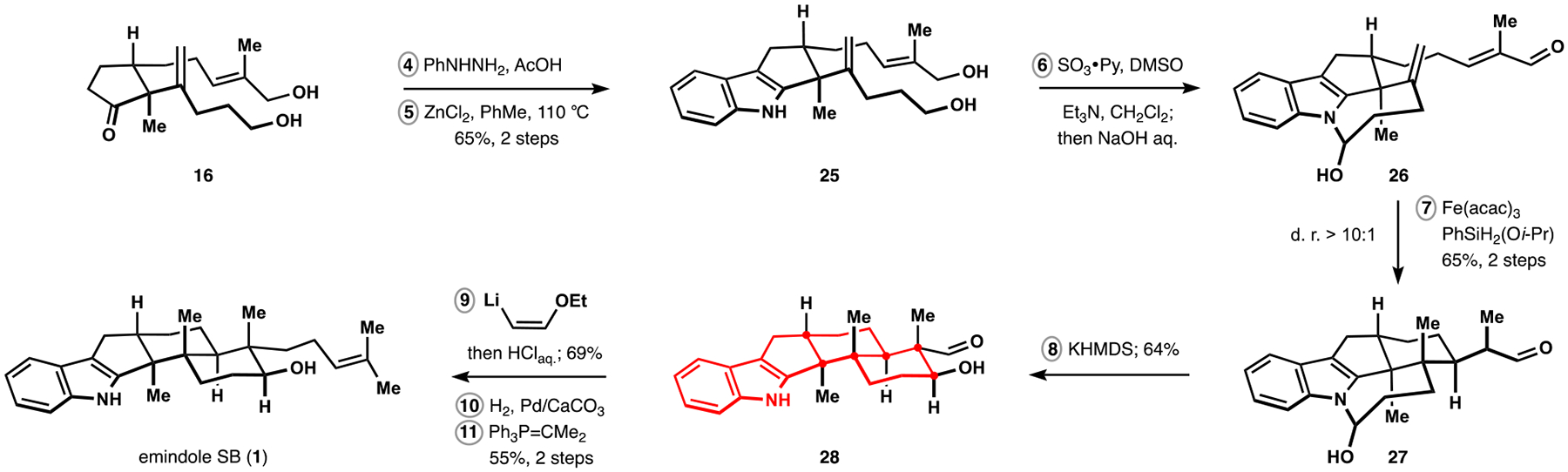
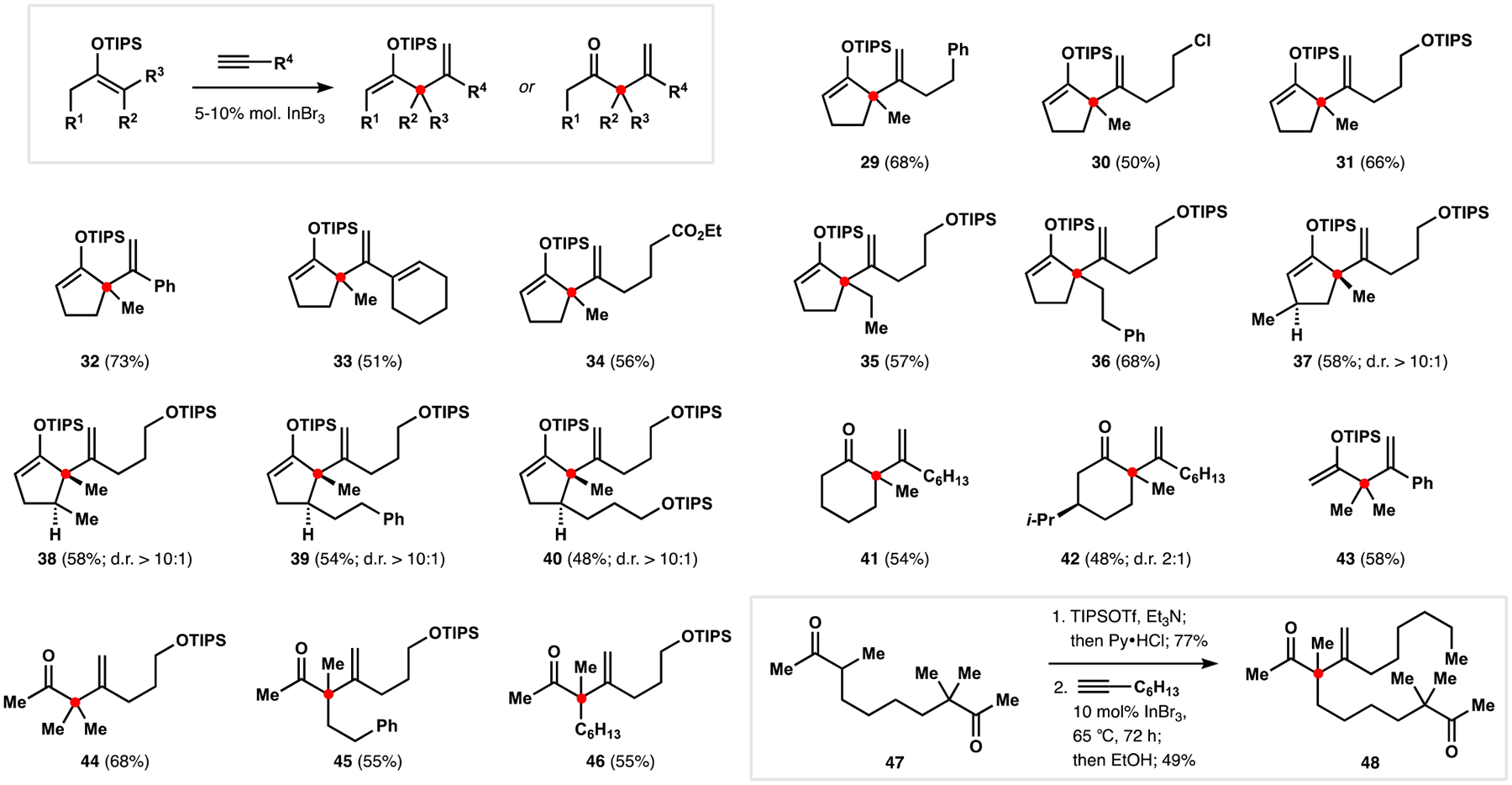
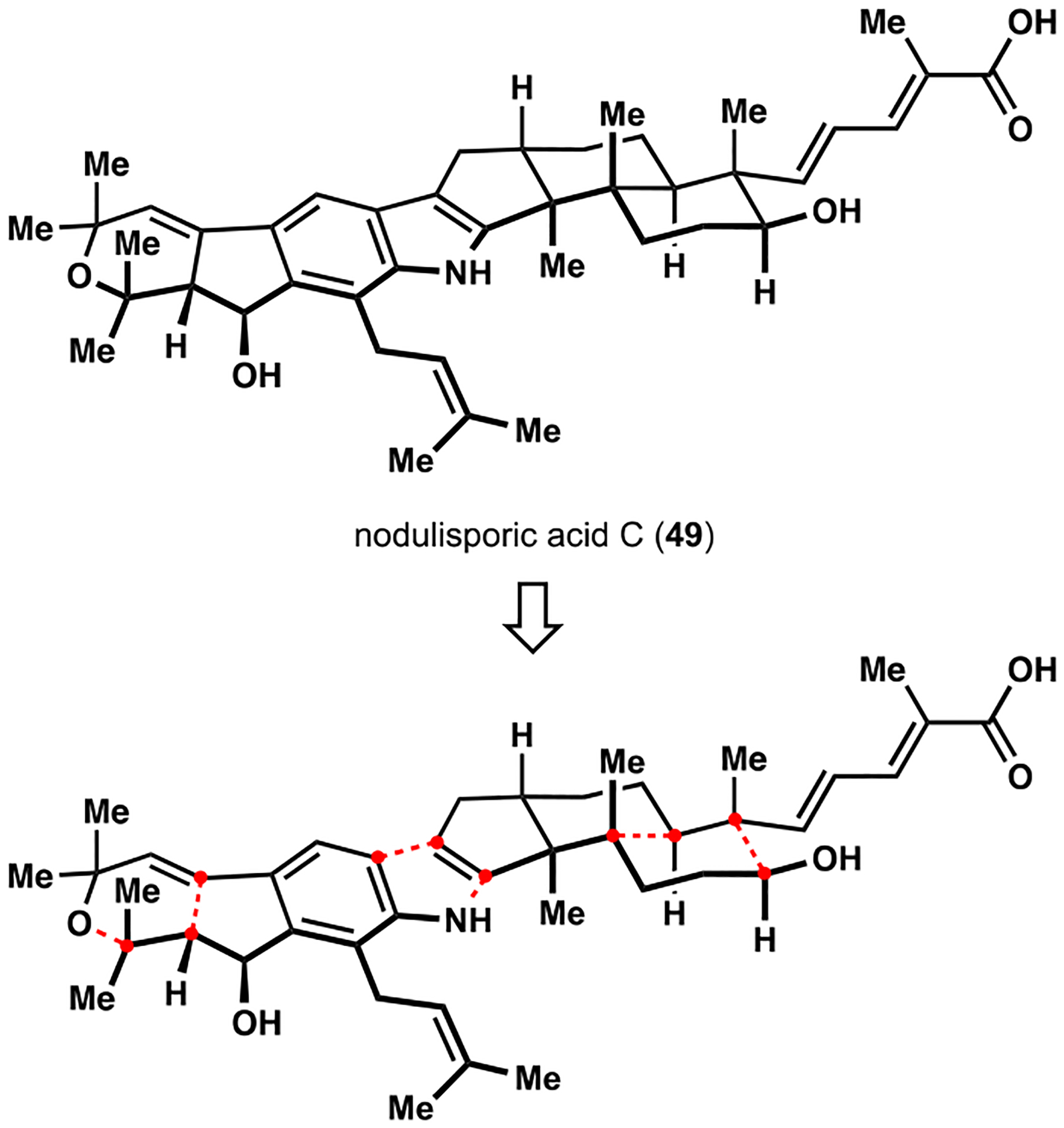
Indoloterpenoids of the paxilline type belong to a large family of secondary metabolites that exhibit unique molecular architectures and a diverse set of biological activities. More than 100 congeners identified to date share a common structural motif that contains an indole moiety fused to a rearranged diterpenoid fragment. The representative physiological and cellular effects attributed to this family of natural products include neurological and insecticidal activities, modulation of lipid balance, and inhibition of mitosis. The uniting polycyclic motif combined with the diversity of individual structural features of paxilline indoloterpenoids and the broad scope of their biological activities have fascinated organic chemists for the past four decades and have led to the development of numerous syntheses. In this Account, we describe our contributions to this field and how they in turn shape new directions that are developing in our laboratory.We begin with the discussion of our strategy for the synthesis of the shared indoloterpenoid core. To address stereochemical challenges encountered in earlier reports, we planned to leverage a suitably substituted cyclopentanone in a polycyclization to form the desired -decalin motif. This polycyclization relied on a radical-polar crossover cascade initiated by hydrogen atom transfer. The original process exhibited poor diastereoselectivity, but we discovered an efficient solution to this problem that took advantage of intramolecular tethering effects, culminating in short synthesis of emindole SB. During these studies, we also identified indium-mediated alkenylation of silyl enol ethers with alkynes as a suitable method for the synthesis of highly substituted β,γ-unsaturated ketones that was critical to achieving brevity of our route. We subsequently developed a catalytic version of this transformation that allowed for a formal bimolecular ene reaction that exhibited unusual and potentially useful selectivity in construction of quaternary centers.To test the scope and limitations of our approach to paxilline indoloterpenoids and identify potential improvements, we developed a synthesis of the more complex congener nodulisporic acid C. The convergent assembly of this natural product was enabled by identification of new elements of stereocontrol in the radical-polar crossover polycyclization en route to the polycyclic terpenoid motif and development of a highly diastereoselective enyne cycloisomerization to access the indenopyran motif and a ketone arylation protocol to unite the two complex fragments.In subsequent studies, we expanded the radical-polar crossover cascade underlying our approach to paxilline indoloterpenoids to a bimolecular setting, which allowed for annulation of two unsaturated carbonyl components to produce functionalized cyclohexanes. This transformation is particularly well suited for installation of fully substituted carbons and can be complementary to the venerable Diels-Alder reaction. The utility of the new annulation was tested in the synthesis of forskolin, allowing for rapid construction of the complex polycyclic motif in this densely functionalized labdane diterpenoid.Over the past five years, our initial forays into the synthesis of paxilline indoloterpenoids have grown into a program that incorporates development of new synthetic methods and pursues artificial assembly of terpenoid natural products from several different families. We are encouraged by the increasing diversity of structural motifs made accessible by application of this chemistry and continue to discover new aspects of the underlying reactivity.
帕西利宁型吲哚萜类化合物属于一大类次生代谢产物,具有独特的分子结构和多样化的生物活性。迄今为止,已经鉴定出 100 多种同系物,它们具有共同的结构母核,包含一个融合了重排二萜片段的吲哚部分。该家族天然产物的代表性生理和细胞效应包括神经和杀虫活性、脂质平衡的调节以及有丝分裂的抑制。这种多环结构基序与帕西利宁吲哚萜类化合物的个体结构特征的多样性以及它们广泛的生物活性相结合,吸引了有机化学家们长达四十年的关注,并导致了许多合成方法的发展。在本综述中,我们描述了我们在这一领域的贡献,以及它们如何为我们实验室正在发展的新方向提供了思路。
我们首先讨论了合成共享吲哚萜类核心的策略。为了解决早期报道中遇到的立体化学挑战,我们计划在多环化反应中利用适当取代的环戊酮来形成所需的 - 去甲环戊烷结构。这种多环化反应依赖于氢原子转移引发的自由基-极性交叉级联。最初的过程表现出较差的非对映选择性,但我们发现了一个有效的解决方案,利用了分子内键合效应,最终实现了 emindole SB 的短合成。在这些研究中,我们还确定了铟介导的硅基烯醇醚与炔烃的烯丙基化反应是合成高度取代的β,γ-不饱和酮的合适方法,这对于实现我们路线的简洁性至关重要。随后,我们开发了该反应的催化版本,允许进行形式上的双分子烯反应,该反应在构建季碳原子方面表现出了不寻常且可能有用的选择性。
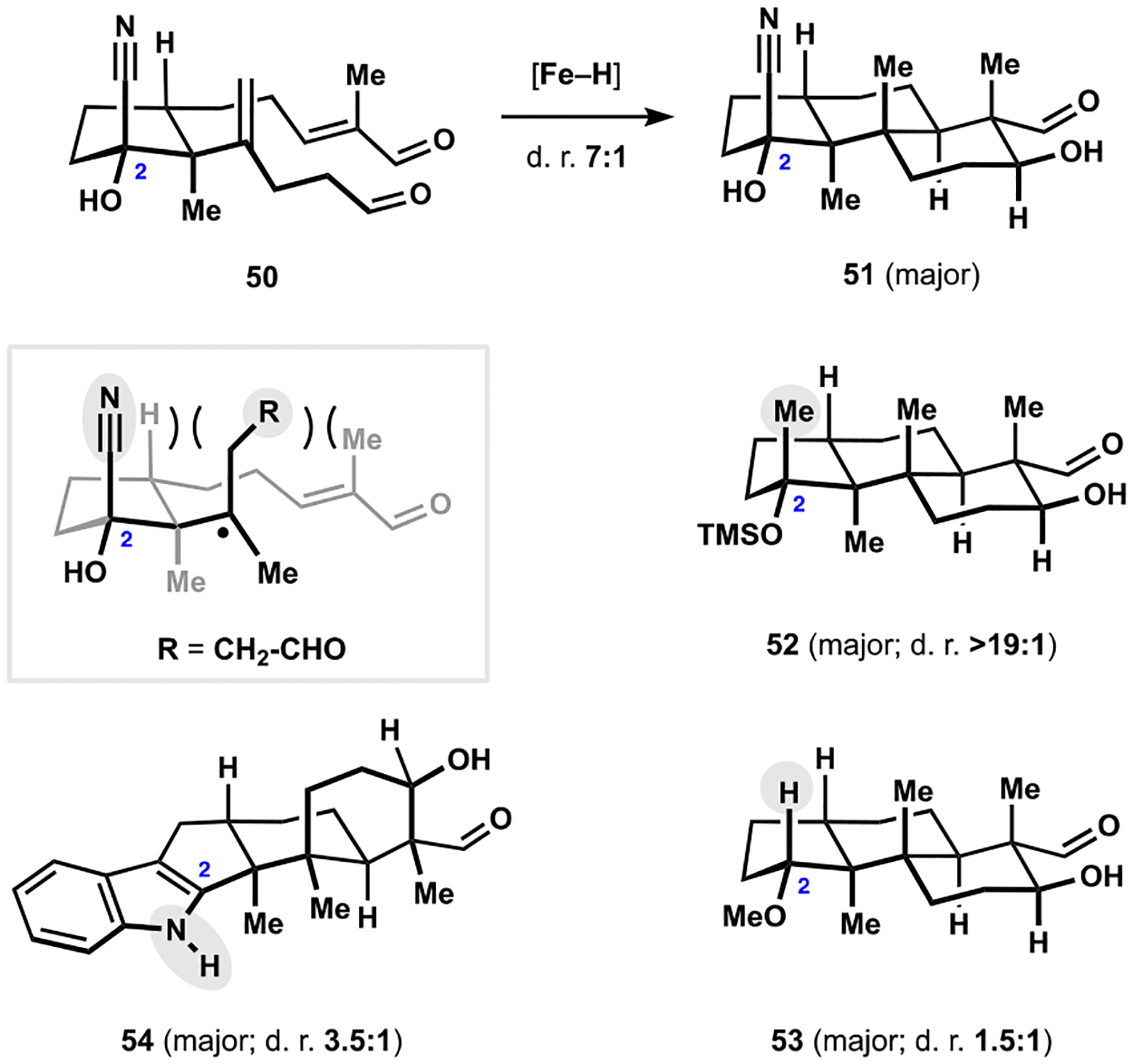
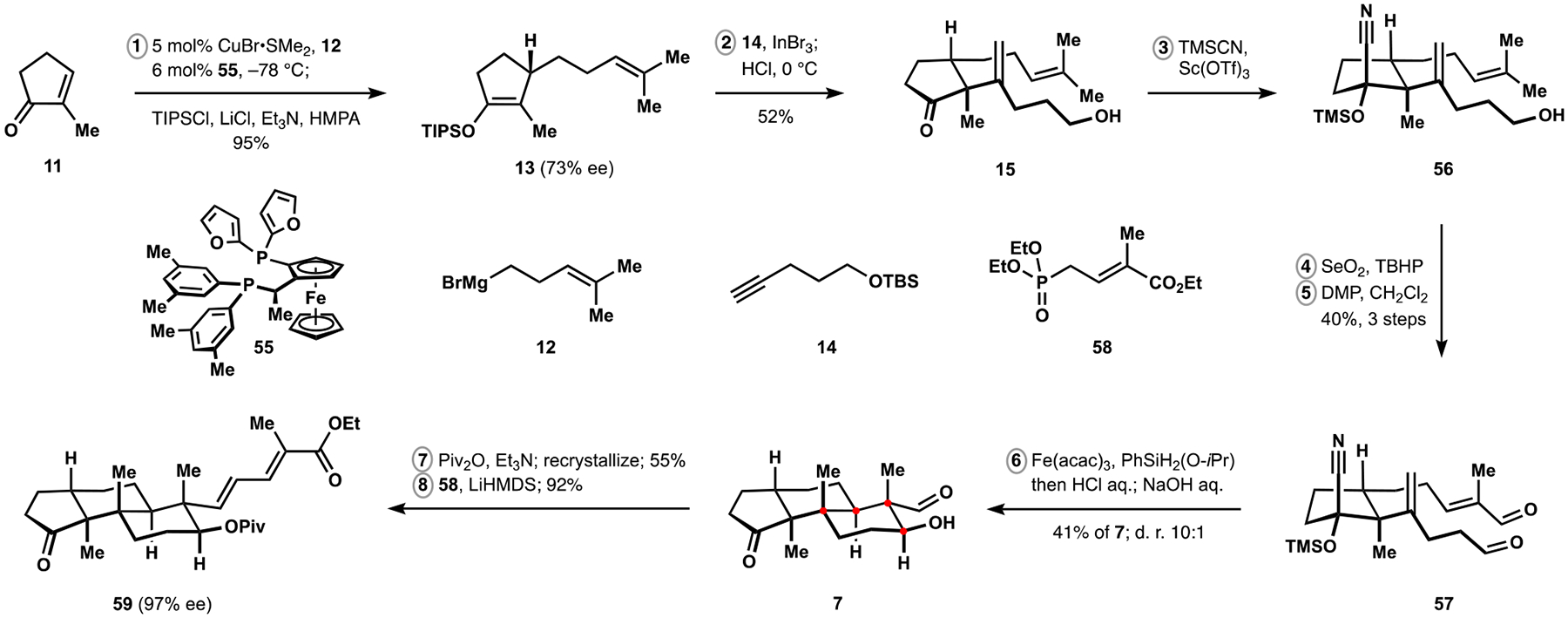
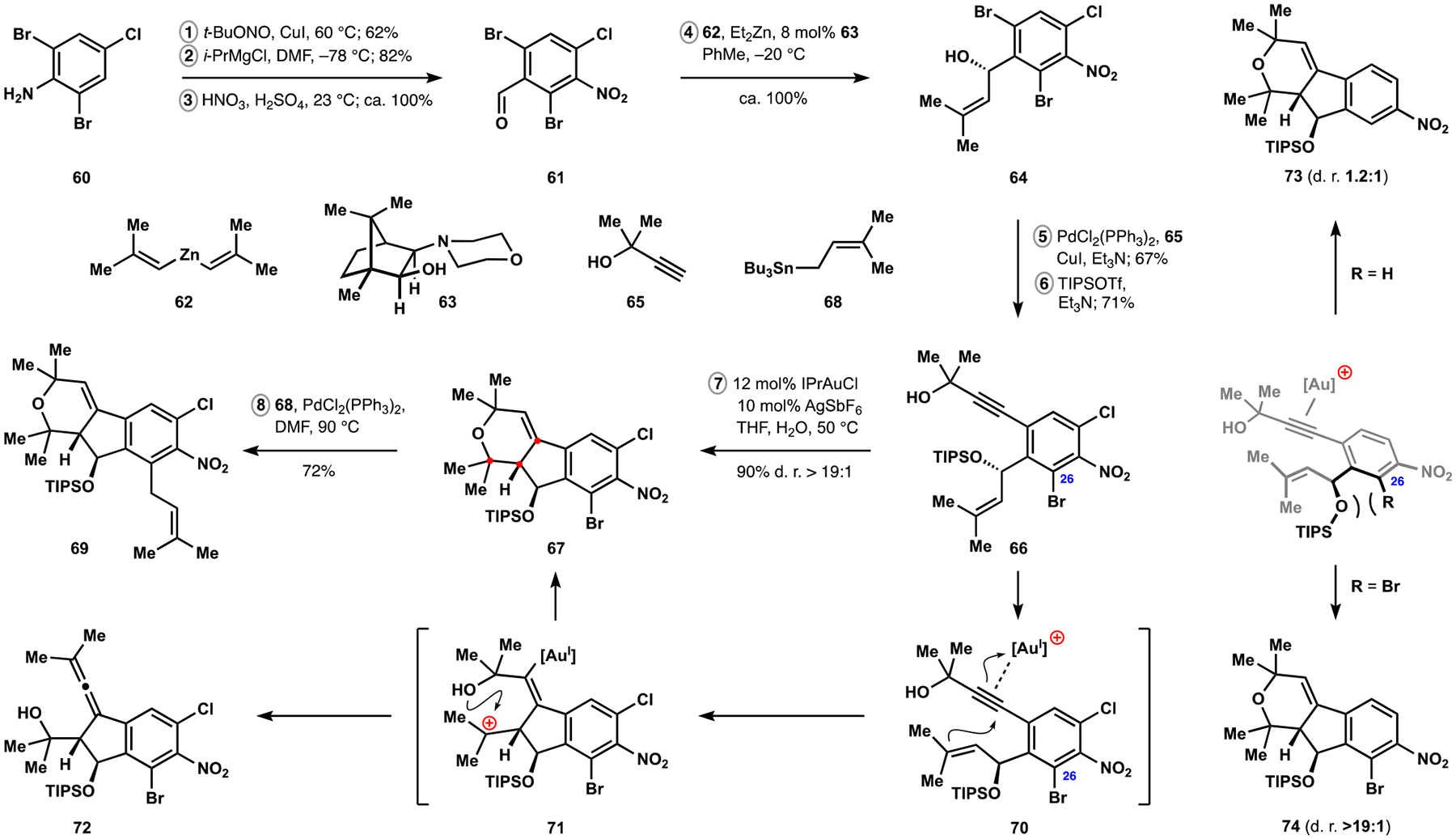
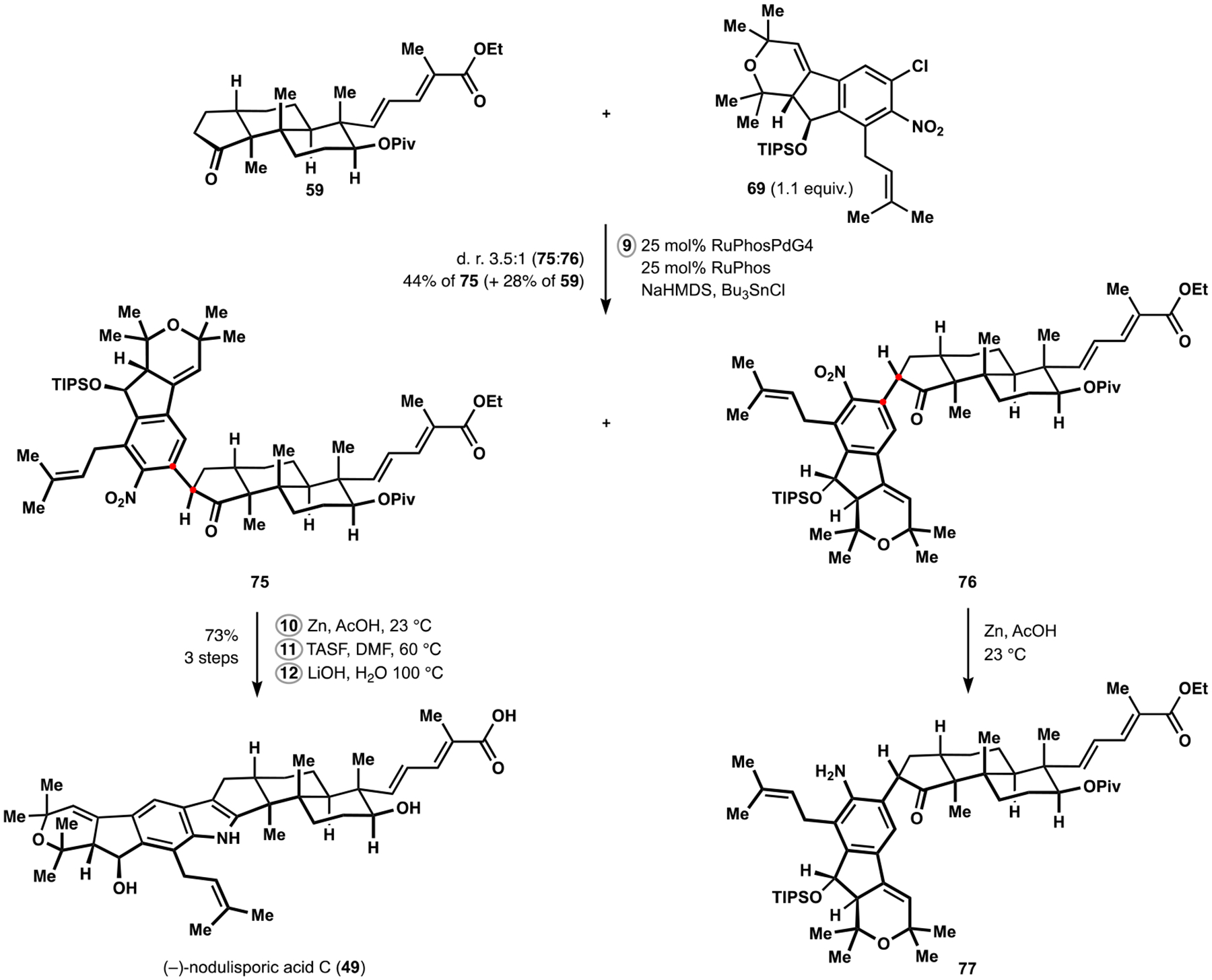
为了测试我们对帕西利宁吲哚萜类化合物的方法的范围和局限性,并确定潜在的改进,我们开发了一种更复杂的同系物 nodulisporic acid C 的合成方法。该天然产物的汇聚组装是通过识别在多环萜烯结构母核的自由基-极性交叉多环化反应中立体控制的新元素以及开发高非对映选择性的烯炔环异构化反应来实现的,该反应可以进入吲哚吡喃结构母核,并开发了酮芳基化反应来连接两个复杂片段。
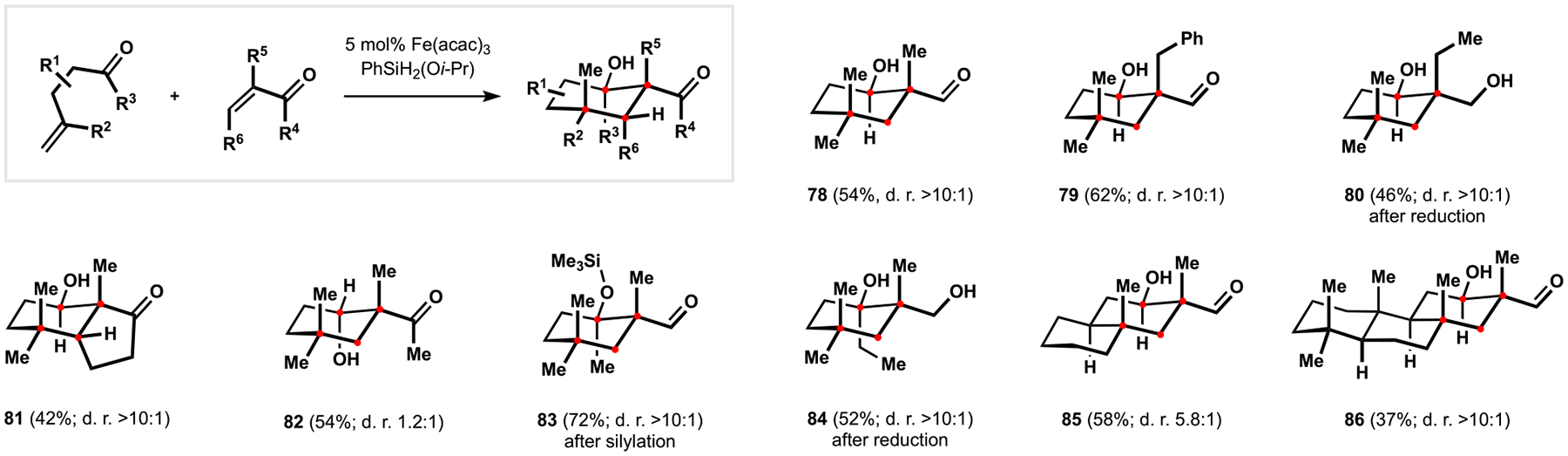
在随后的研究中,我们将我们的帕西利宁吲哚萜类化合物方法中的自由基-极性交叉级联扩展到双分子体系中,这允许两个不饱和羰基成分的环加成反应生成功能化的环己烷。这种转化特别适合于完全取代的碳的安装,并且可以与古老的 Diels-Alder 反应互补。新环加成反应的实用性在 forskolin 的合成中得到了测试,允许在这个高度功能化的 labdane 二萜中快速构建复杂的多环结构母核。
在过去的五年中,我们最初对帕西利宁吲哚萜类化合物的合成探索已经发展成为一个项目,该项目包括开发新的合成方法,并从几个不同的家族中追求萜类天然产物的人工组装。我们对应用这种化学方法所获得的结构基序的多样性感到鼓舞,并继续发现其基础反应的新方面。