Ruiz-Perez Daniel, Lugo-Martinez Jose, Bourguignon Natalia, Mathee Kalai, Lerner Betiana, Bar-Joseph Ziv, Narasimhan Giri
Florida International University, Bioinformatics Research Group (BioRG), Miami, Florida, USA.
Carnegie Mellon University, Computational Biology Department, School of Computer Science, Pittsburgh, Pennsylvania, USA.
mSystems. 2021 Mar 30;6(2):e01105-20. doi: 10.1128/mSystems.01105-20.
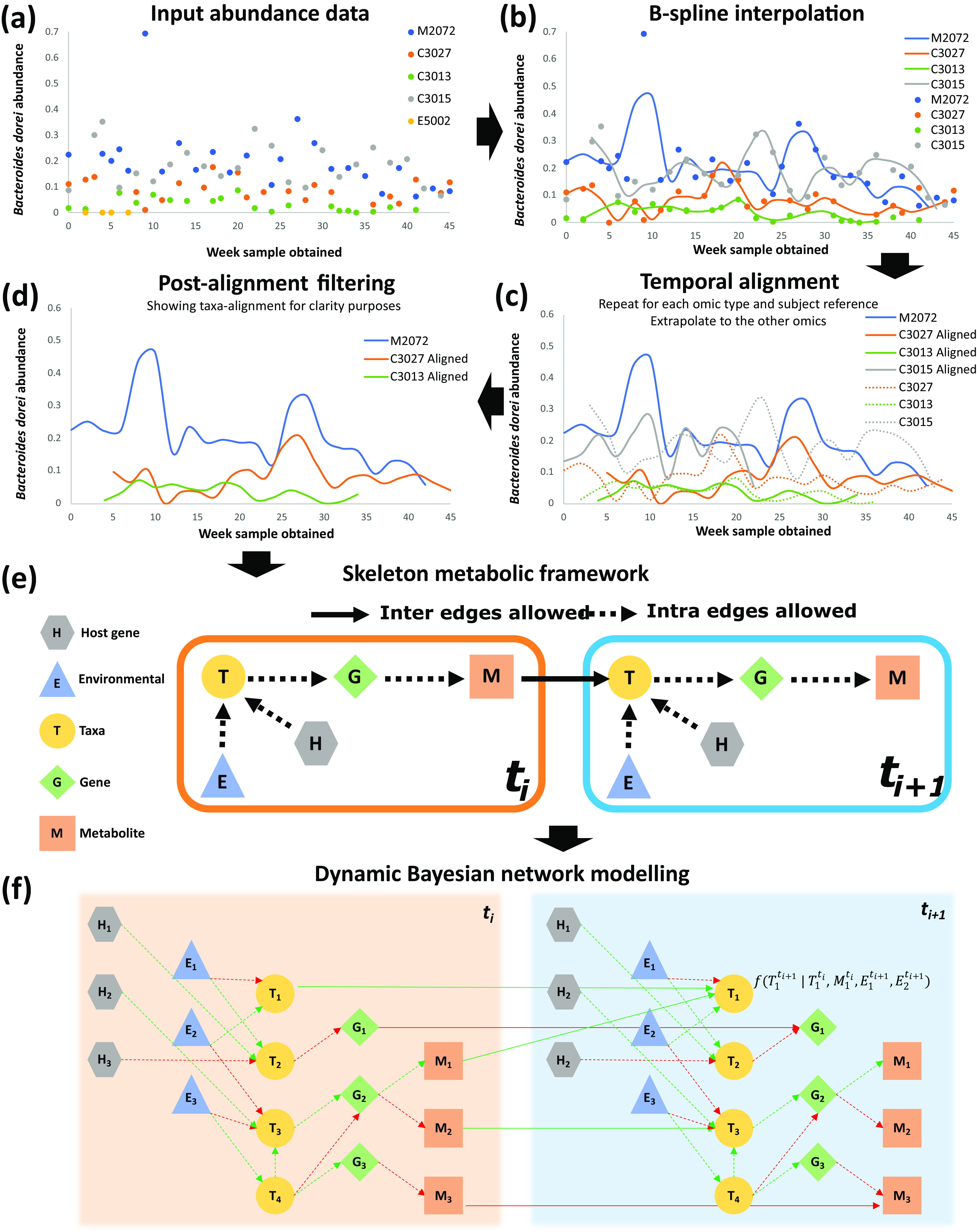
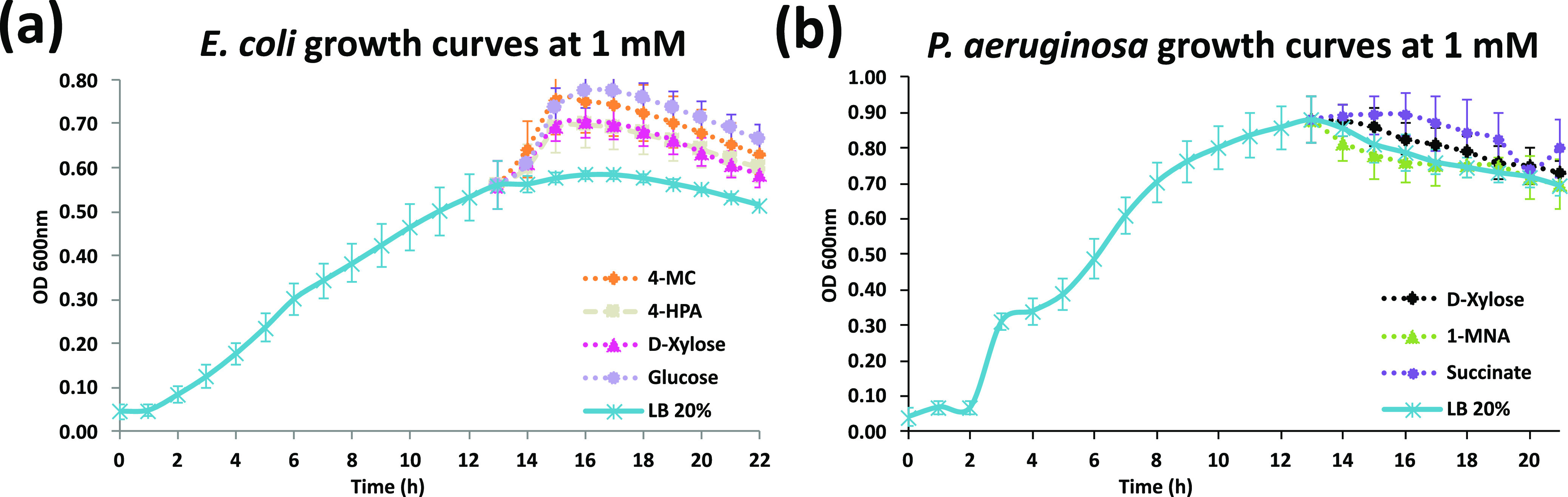
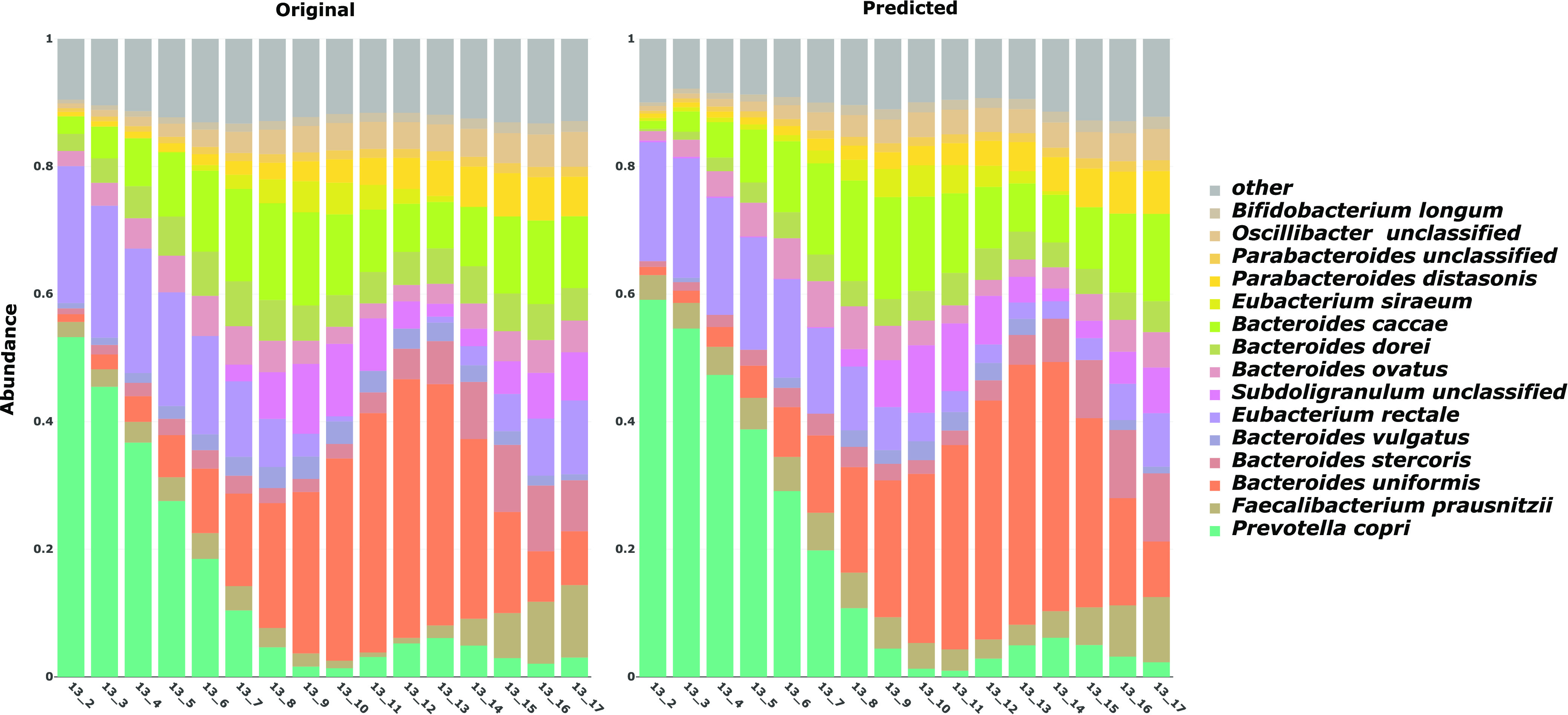
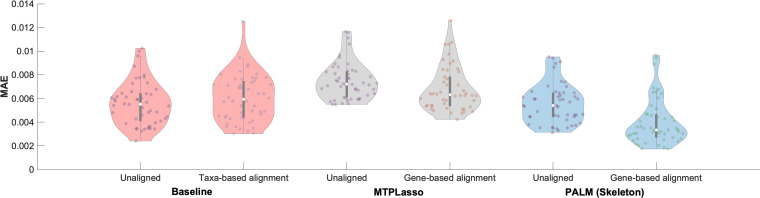
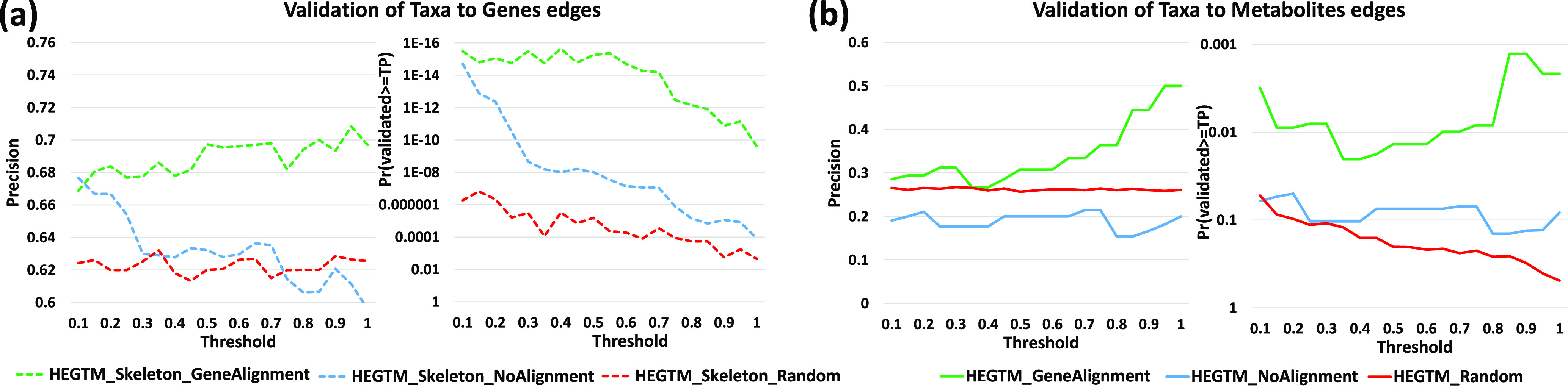
A key challenge in the analysis of longitudinal microbiome data is the inference of temporal interactions between microbial taxa, their genes, the metabolites that they consume and produce, and host genes. To address these challenges, we developed a computational pipeline, a pipeline for the analysis of longitudinal multi-omics data (PALM), that first aligns multi-omics data and then uses dynamic Bayesian networks (DBNs) to reconstruct a unified model. Our approach overcomes differences in sampling and progression rates, utilizes a biologically inspired multi-omic framework, reduces the large number of entities and parameters in the DBNs, and validates the learned network. Applying PALM to data collected from inflammatory bowel disease patients, we show that it accurately identifies known and novel interactions. Targeted experimental validations further support a number of the predicted novel metabolite-taxon interactions. While a number of large consortia collect and profile several different types of microbiome and genomic time series data, very few methods exist for joint modeling of multi-omics data sets. We developed a new computational pipeline, PALM, which uses dynamic Bayesian networks (DBNs) and is designed to integrate multi-omics data from longitudinal microbiome studies. When used to integrate sequence, expression, and metabolomics data from microbiome samples along with host expression data, the resulting models identify interactions between taxa, their genes, and the metabolites that they produce and consume, as well as their impact on host expression. We tested the models both by using them to predict future changes in microbiome levels and by comparing the learned interactions to known interactions in the literature. Finally, we performed experimental validations for a few of the predicted interactions to demonstrate the ability of the method to identify novel relationships and their impact.
纵向微生物组数据分析中的一个关键挑战是推断微生物分类群、它们的基因、它们消耗和产生的代谢物以及宿主基因之间的时间相互作用。为了应对这些挑战,我们开发了一种计算流程,即纵向多组学数据分析流程(PALM),该流程首先对多组学数据进行比对,然后使用动态贝叶斯网络(DBN)来重建一个统一模型。我们的方法克服了采样和进展速率方面的差异,利用了受生物学启发的多组学框架,减少了DBN中大量的实体和参数,并对学习到的网络进行了验证。将PALM应用于从炎症性肠病患者收集的数据,我们表明它能够准确识别已知和新的相互作用。有针对性的实验验证进一步支持了一些预测的新的代谢物-分类群相互作用。虽然有一些大型联盟收集并分析了几种不同类型的微生物组和基因组时间序列数据,但用于多组学数据集联合建模的方法却非常少。我们开发了一种新的计算流程PALM,它使用动态贝叶斯网络(DBN),旨在整合纵向微生物组研究中的多组学数据。当用于整合微生物组样本的序列、表达和代谢组学数据以及宿主表达数据时,生成的模型能够识别分类群、它们的基因以及它们产生和消耗的代谢物之间的相互作用,以及它们对宿主表达的影响。我们通过使用这些模型预测微生物组水平的未来变化以及将学习到的相互作用与文献中的已知相互作用进行比较来测试这些模型。最后,我们对一些预测的相互作用进行了实验验证,以证明该方法识别新关系及其影响的能力。