Liu Dan, Qu Kai, Yuan Yangchen, Zhao Zhiheng, Chen Ying, Han Biao, Li Wei, El-Kassaby Yousry A, Yin Yangyang, Xie Xiaoman, Tong Boqiang, Liu Hongshan
Shandong Provincial Center of Forest and Grass Germplasm Resources, Jinan, China.
State Key Laboratory of Tree Genetics and Breeding, National Engineering Research Center of Tree Breeding and Ecological Restoration, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, China.
Front Genet. 2023 Jan 4;13:1050040. doi: 10.3389/fgene.2022.1050040. eCollection 2022.
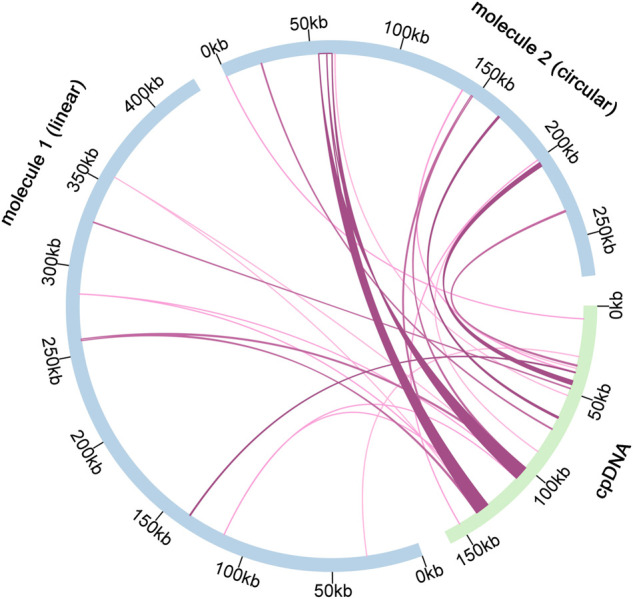
is one of the large worldwide genera of the Ranunculaceae Juss. Family, with high ornamental and medicinal value. China is the modern distribution centre of with abundant natural populations. Due to the complexity and high morphological diversity of , the genus is difficult to classify systematically, and in particular, the phylogenetic position of the endangered is highly controversial. The use of the mitochondrial complete genome is a powerful molecular method that is frequently used for inferring plants phylogenies. However, studies on mitogenome are rare, thus limiting our full understanding of its phylogeny and genome evolution. Here, we sequenced and annotated the mt genome using Illumina short- and Nanopore long-reads, characterized the species first complete mitogenome, and performed a comparative phylogenetic analysis with its close relatives. The total length of the mitogenome is 698,247 bp and the main structure is multi-branched (linear molecule 1 and circular molecule 2). We annotated 55 genes, including 35 protein-coding, 17 tRNA, and 3 rRNA genes. The mitogenome has extremely unconserved structurally, with extensive sequence transfer between the chloroplast and mitochondrial organelles, sequence repeats, and RNA editing. The phylogenetic position of was determined by constructing the species mitogenome with 24 angiosperms. Further, our mitogenome characteristics investigation included GC contents, codon usage, repeats and synteny analysis. Overall, our results are expected to provide fundamental information for mitogenome evolution and confirm the validity of mitochondrial analysis in determining the phylogenetic positioning of plants.
是毛茛科(Juss.)全球较大的属之一,具有很高的观赏和药用价值。中国是其现代分布中心,有丰富的自然种群。由于该属植物形态复杂且多样性高,难以进行系统分类,尤其是濒危物种的系统发育位置极具争议。线粒体全基因组的使用是一种强大的分子方法,常用于推断植物系统发育。然而,关于该属线粒体基因组的研究很少,这限制了我们对其系统发育和基因组进化的全面理解。在此,我们利用Illumina短读长和Nanopore长读长对该属线粒体基因组进行测序和注释,表征了该物种的首个完整线粒体基因组,并与其近缘种进行了比较系统发育分析。该属线粒体基因组全长698,247 bp,主要结构为多分支(线性分子1和环状分子2)。我们注释了55个基因,包括35个蛋白质编码基因、17个tRNA基因和3个rRNA基因。该属线粒体基因组在结构上极不保守,叶绿体和线粒体细胞器之间存在广泛的序列转移、序列重复和RNA编辑。通过构建该物种线粒体基因组与24种被子植物的关系确定了其系统发育位置。此外,我们对该属线粒体基因组特征的研究包括GC含量、密码子使用、重复序列和共线性分析。总体而言,我们的研究结果有望为该属线粒体基因组进化提供基础信息,并证实线粒体分析在确定该属植物系统发育定位中的有效性。