Zhao Shu, Ge Wenbo, Watanabe Akira, Fortwendel Jarrod R, Gibbons John G
Molecular and Cellular Biology Graduate Program, University of Massachusetts, Amherst, MA, United States.
Department of Food Science, University of Massachusetts, Amherst, MA, United States.
Front Fungal Biol. 2021 Jan 14;1:617338. doi: 10.3389/ffunb.2020.617338. eCollection 2020.
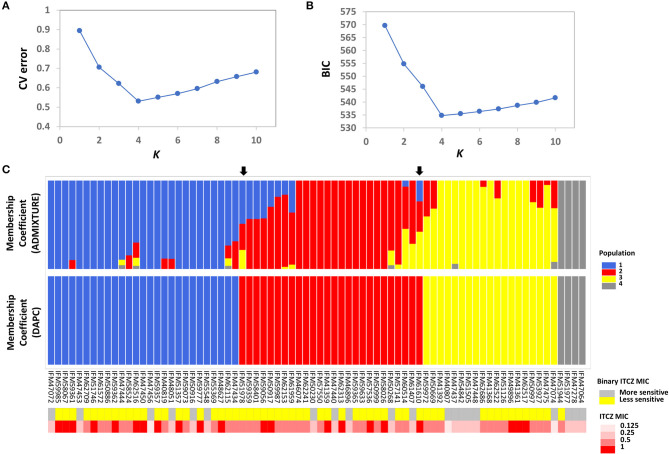
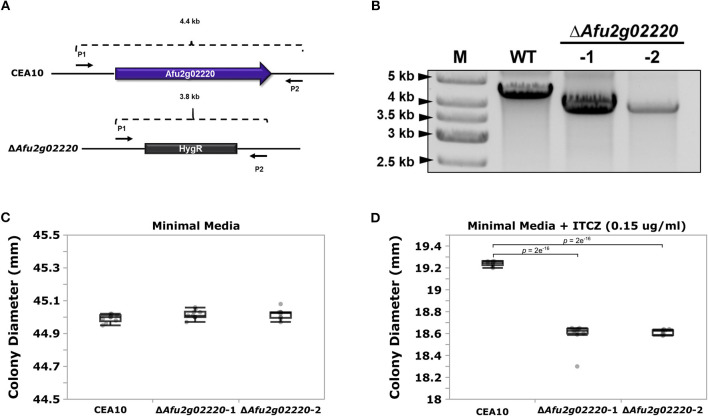
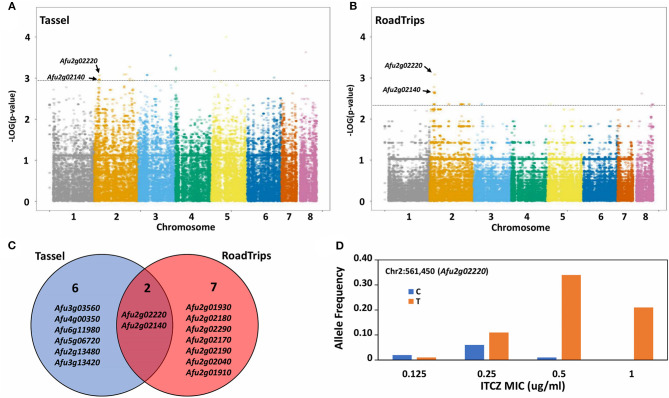
is a potentially lethal opportunistic pathogen that infects over ~200,000 people and causes ~100,000 deaths per year globally. Treating infections is particularly challenging because of the recent emergence of azole-resistance. The majority of studies focusing on the molecular mechanisms underlying azole resistance have examined azole-resistant isolates. However, isolates that are susceptible to azoles also display variation in their sensitivity, presenting a unique opportunity to identify genes contributing to azole sensitivity. Here, we used genome-wide association (GWA) analysis to identify loci involved in azole sensitivity by analyzing the association between 68,853 SNPs and itraconazole (ITCZ) minimum inhibitory concentration (MIC) in 76 clinical isolates of from Japan. Population structure analysis suggests the presence of four distinct populations, with ITCZ MICs distributed relatively evenly across populations. We independently conducted GWA when treating ITCZ MIC as a quantitative trait and a binary trait, and identified two SNPs with strong associations in both analyses. These SNPs fell within the coding regions of and . We functionally validated by knocking it out using a CRISPR/Cas9 approach, because orthologs of this gene are involved in sterol modification and ITCZ targets the ergosterol biosynthesis pathway. Knockout strains displayed no difference in growth compared to the parent strain in minimal media, yet a minor but consistent inhibition of growth in the presence of 0.15 μg/ml ITCZ. Our results suggest that GWA paired with efficient gene deletion is a powerful and unbiased strategy for identifying the genetic basis of complex traits in .
是一种潜在致命的机会性病原体,全球每年感染约20万人并导致约10万例死亡。由于最近出现了唑类耐药性,治疗感染尤其具有挑战性。大多数关注唑类耐药性分子机制的研究都检查了唑类耐药菌株。然而,对唑类敏感的菌株在敏感性上也存在差异,这为鉴定影响唑类敏感性的基因提供了独特机会。在这里,我们通过分析来自日本的76株临床分离株中68,853个单核苷酸多态性(SNP)与伊曲康唑(ITCZ)最低抑菌浓度(MIC)之间的关联,利用全基因组关联(GWA)分析来鉴定与唑类敏感性相关的基因座。群体结构分析表明存在四个不同的群体,ITCZ MIC在各群体中分布相对均匀。我们在将ITCZ MIC作为定量性状和二元性状处理时分别进行了GWA分析,并在两项分析中均鉴定出两个具有强关联的SNP。这些SNP位于 和 的编码区域内。我们通过使用CRISPR/Cas9方法敲除 进行了功能验证,因为该基因的直系同源物参与甾醇修饰,而ITCZ靶向麦角甾醇生物合成途径。与亲本菌株相比,敲除菌株在基本培养基中的生长没有差异,但在存在0.15μg/ml ITCZ时生长受到轻微但持续的抑制。我们的结果表明,GWA与高效基因缺失相结合是一种强大且无偏倚的策略,可用于鉴定 的复杂性状的遗传基础。