Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, PA, USA.
Penn Ovarian Cancer Research Center, Department of Obstetrics and Gynecology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA.
Genome Biol. 2023 Oct 20;24(1):239. doi: 10.1186/s13059-023-03077-7.
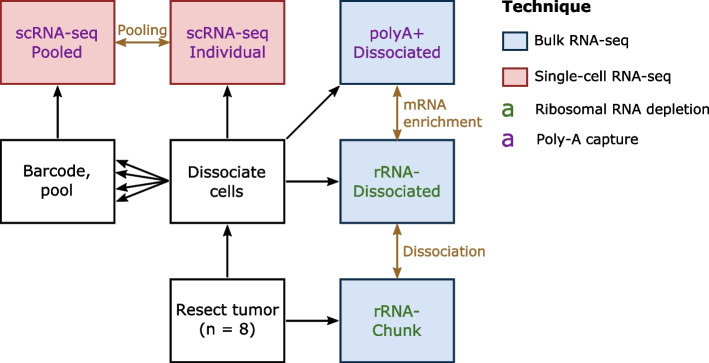
Single-cell gene expression profiling provides unique opportunities to understand tumor heterogeneity and the tumor microenvironment. Because of cost and feasibility, profiling bulk tumors remains the primary population-scale analytical strategy. Many algorithms can deconvolve these tumors using single-cell profiles to infer their composition. While experimental choices do not change the true underlying composition of the tumor, they can affect the measurements produced by the assay.
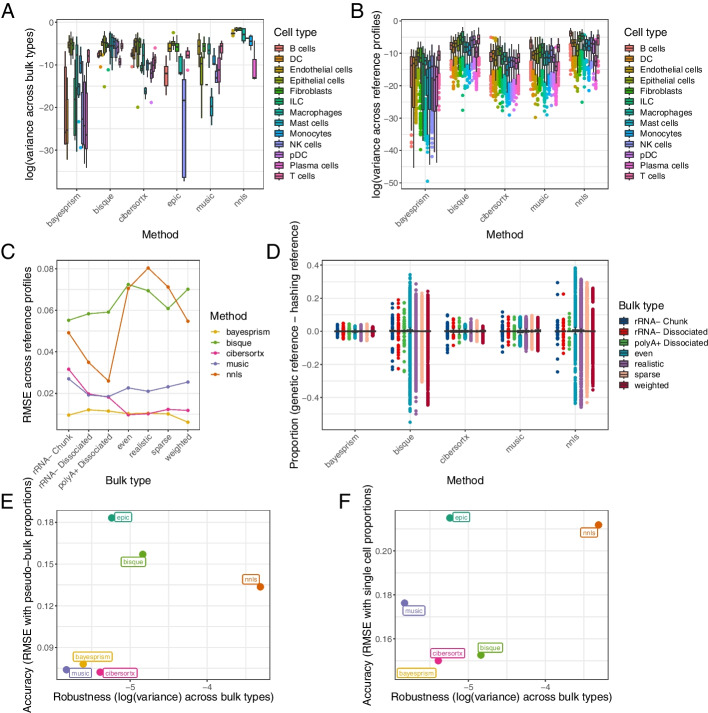
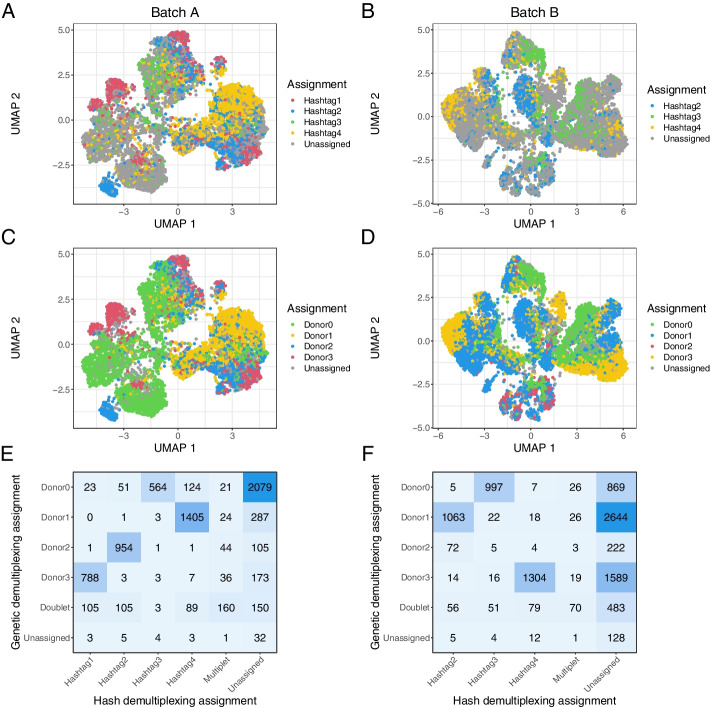
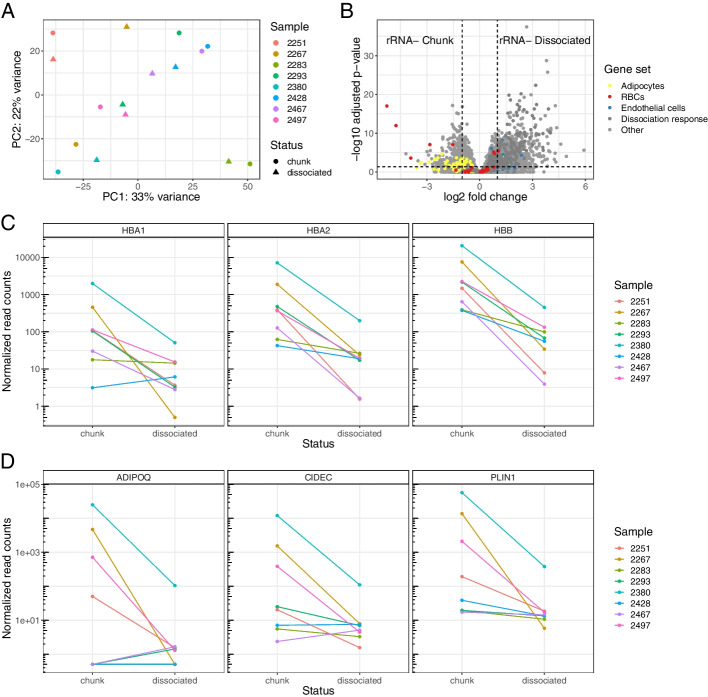
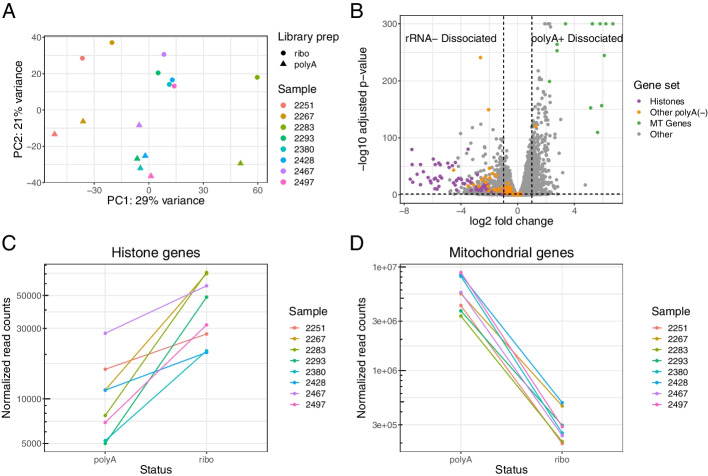
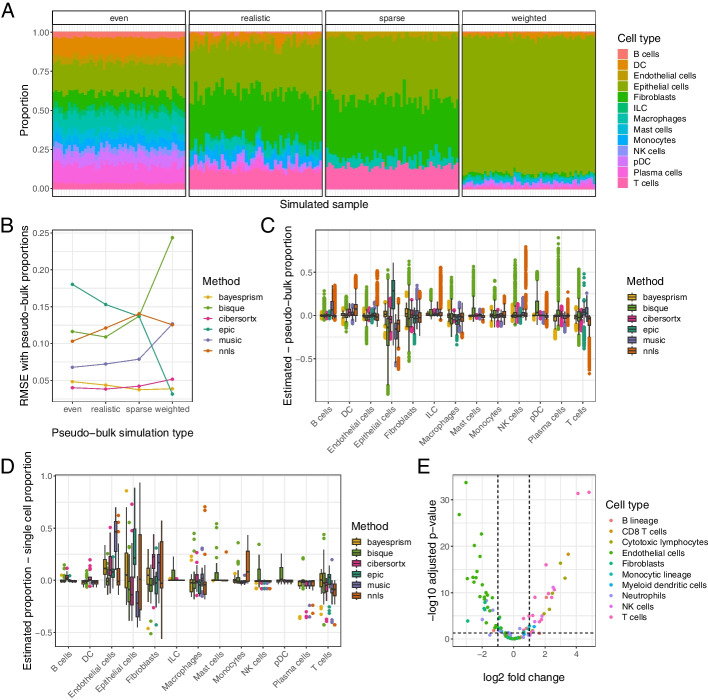
We generated a dataset of high-grade serous ovarian tumors with paired expression profiles from using multiple strategies to examine the extent to which experimental factors impact the results of downstream tumor deconvolution methods. We find that pooling samples for single-cell sequencing and subsequent demultiplexing has a minimal effect. We identify dissociation-induced differences that affect cell composition, leading to changes that may compromise the assumptions underlying some deconvolution algorithms. We also observe differences across mRNA enrichment methods that introduce additional discrepancies between the two data types. We also find that experimental factors change cell composition estimates and that the impact differs by method.
Previous benchmarks of deconvolution methods have largely ignored experimental factors. We find that methods vary in their robustness to experimental factors. We provide recommendations for methods developers seeking to produce the next generation of deconvolution approaches and for scientists designing experiments using deconvolution to study tumor heterogeneity.
单细胞基因表达谱分析为了解肿瘤异质性和肿瘤微环境提供了独特的机会。由于成本和可行性,分析批量肿瘤仍然是主要的群体分析策略。许多算法可以使用单细胞谱来推断其组成,从而对这些肿瘤进行去卷积。虽然实验选择不会改变肿瘤的真实潜在组成,但它们会影响测定产生的测量值。
我们生成了一组高级别浆液性卵巢肿瘤数据集,使用多种策略生成配对的表达谱,以研究实验因素在多大程度上影响下游肿瘤去卷积方法的结果。我们发现,对单细胞测序进行样本合并和随后的多路复用对结果的影响最小。我们确定了分离诱导的差异,这些差异会影响细胞组成,从而导致可能破坏某些去卷积算法假设的变化。我们还观察到不同的 mRNA 富集方法之间存在差异,这会导致两种数据类型之间产生额外的差异。我们还发现,实验因素会改变细胞组成估计值,并且方法的影响也不同。
以前的去卷积方法基准测试在很大程度上忽略了实验因素。我们发现,方法在对实验因素的稳健性方面存在差异。我们为寻求开发下一代去卷积方法的方法开发人员以及为使用去卷积研究肿瘤异质性的科学家设计实验提供了建议。