Department of Medicinal Chemistry, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran.
Medicinal and Natural Products Chemistry Research Center, Shiraz University of Medical Sciences, Shiraz, Iran.
Sci Rep. 2024 Apr 2;14(1):7749. doi: 10.1038/s41598-024-58485-x.
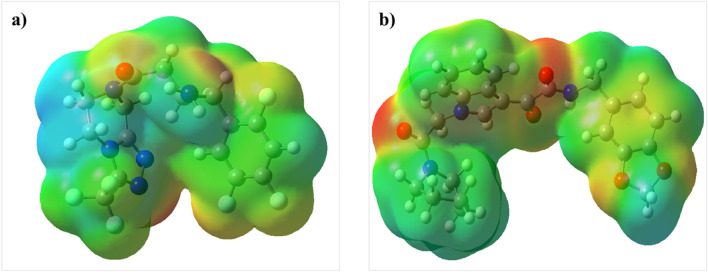
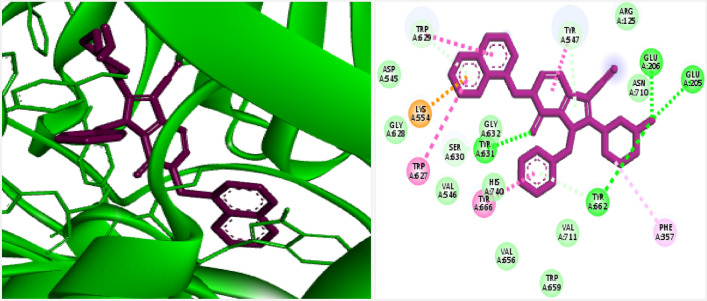
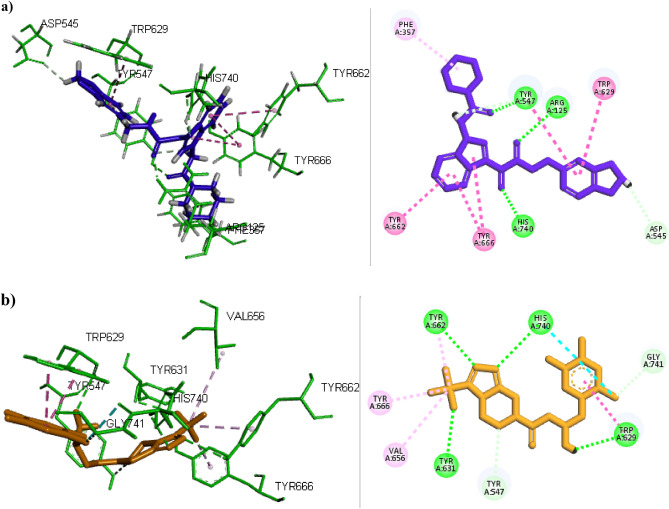
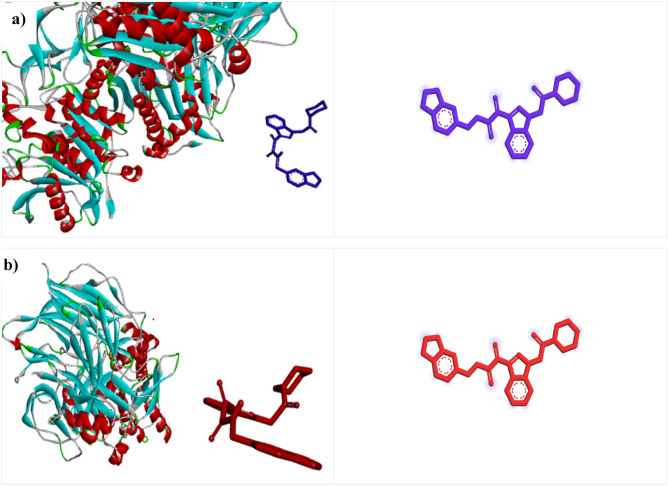
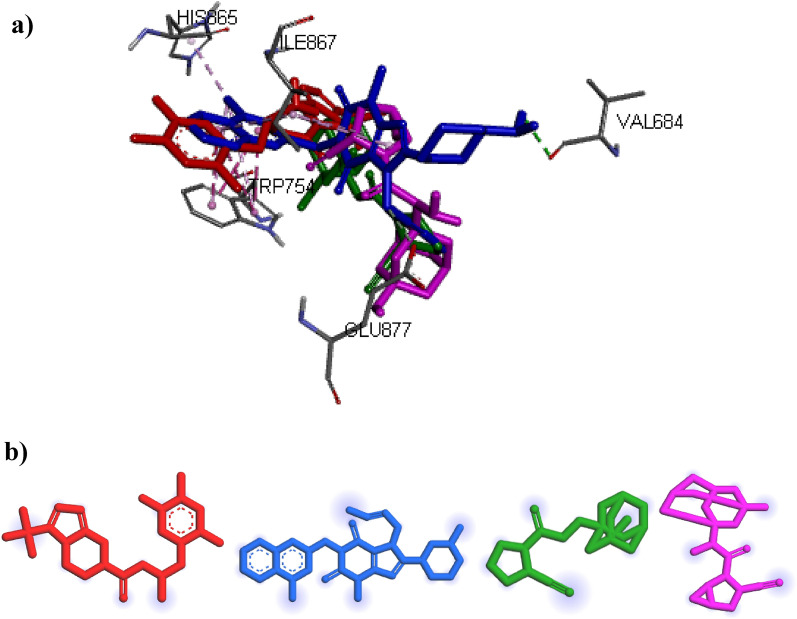
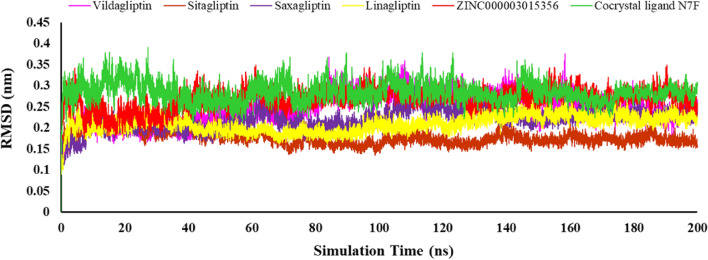
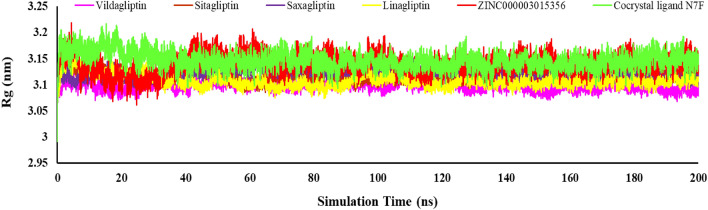
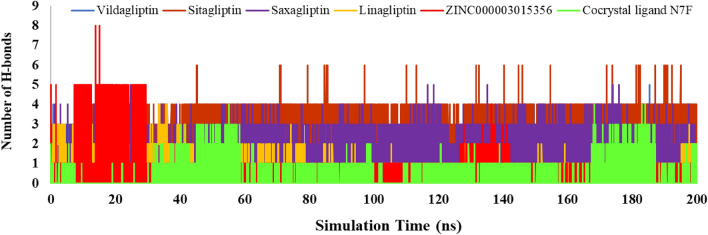
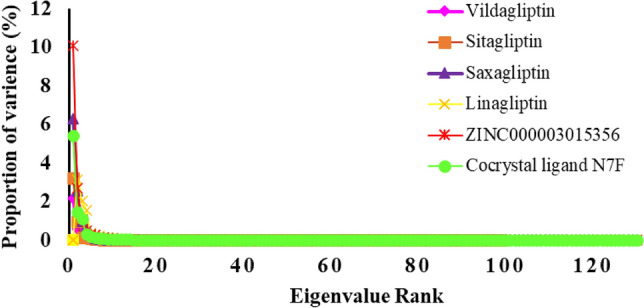
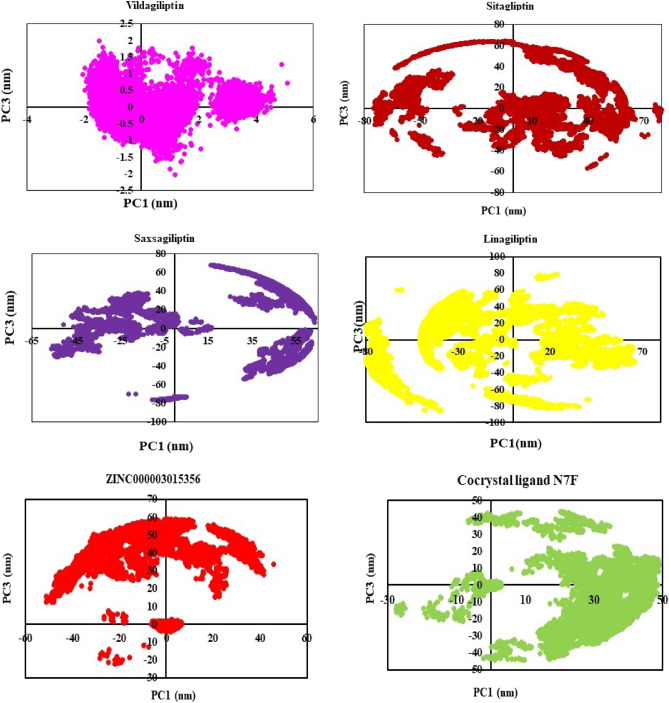
DPP4 inhibitors can control glucose homeostasis by increasing the level of GLP-1 incretins hormone due to dipeptidase mimicking. Despite the potent effects of DPP4 inhibitors, these compounds cause unwanted toxicity attributable to their effect on other enzymes. As a result, it seems essential to find novel and DPP4 selective compounds. In this study, we introduce a potent and selective DPP4 inhibitor via structure-based virtual screening, molecular docking, molecular dynamics simulation, MM/PBSA calculations, DFT analysis, and ADMET profile. The screened compounds based on similarity with FDA-approved DPP4 inhibitors were docked towards the DPP4 enzyme. The compound with the highest docking score, ZINC000003015356, was selected. For further considerations, molecular docking studies were performed on selected ligands and FDA-approved drugs for DPP8 and DPP9 enzymes. Molecular dynamics simulation was run during 200 ns and the analysis of RMSD, RMSF, Rg, PCA, and hydrogen bonding were performed. The MD outputs showed stability of the ligand-protein complex compared to available drugs in the market. The total free binding energy obtained for the proposed DPP4 inhibitor was more negative than its co-crystal ligand (N7F). ZINC000003015356 confirmed the role of the five Lipinski rule and also, have low toxicity parameter according to properties. Finally, DFT calculations indicated that this compound is sufficiently soft.
DPP4 抑制剂可以通过模拟二肽酶来增加 GLP-1 肠促胰岛素激素的水平,从而控制血糖稳态。尽管 DPP4 抑制剂具有强大的作用,但这些化合物会因对其他酶的作用而引起不必要的毒性。因此,似乎有必要寻找新的、具有 DPP4 选择性的化合物。在这项研究中,我们通过基于结构的虚拟筛选、分子对接、分子动力学模拟、MM/PBSA 计算、DFT 分析和 ADMET 分析,介绍了一种有效的、具有选择性的 DPP4 抑制剂。基于与 FDA 批准的 DPP4 抑制剂的相似性,筛选出的化合物与 DPP4 酶对接。对接得分最高的化合物 ZINC000003015356 被选中。为了进一步考虑,对选定的配体和 FDA 批准的 DPP8 和 DPP9 酶抑制剂进行了分子对接研究。进行了 200 ns 的分子动力学模拟,分析了 RMSD、RMSF、Rg、PCA 和氢键。MD 输出表明,与市场上现有的药物相比,配体-蛋白复合物更稳定。与共晶配体(N7F)相比,所提出的 DPP4 抑制剂的总自由结合能更负。ZINC000003015356 证实了五个 Lipinski 规则的作用,并且根据性质具有低毒性参数。最后,DFT 计算表明该化合物足够柔软。