Bioinformatics Research Group , University of Applied Sciences Upper Austria , Softwarepark 11 , 4232 Hagenberg , Austria.
J Proteome Res. 2018 Aug 3;17(8):2581-2589. doi: 10.1021/acs.jproteome.7b00836. Epub 2018 Jun 28.
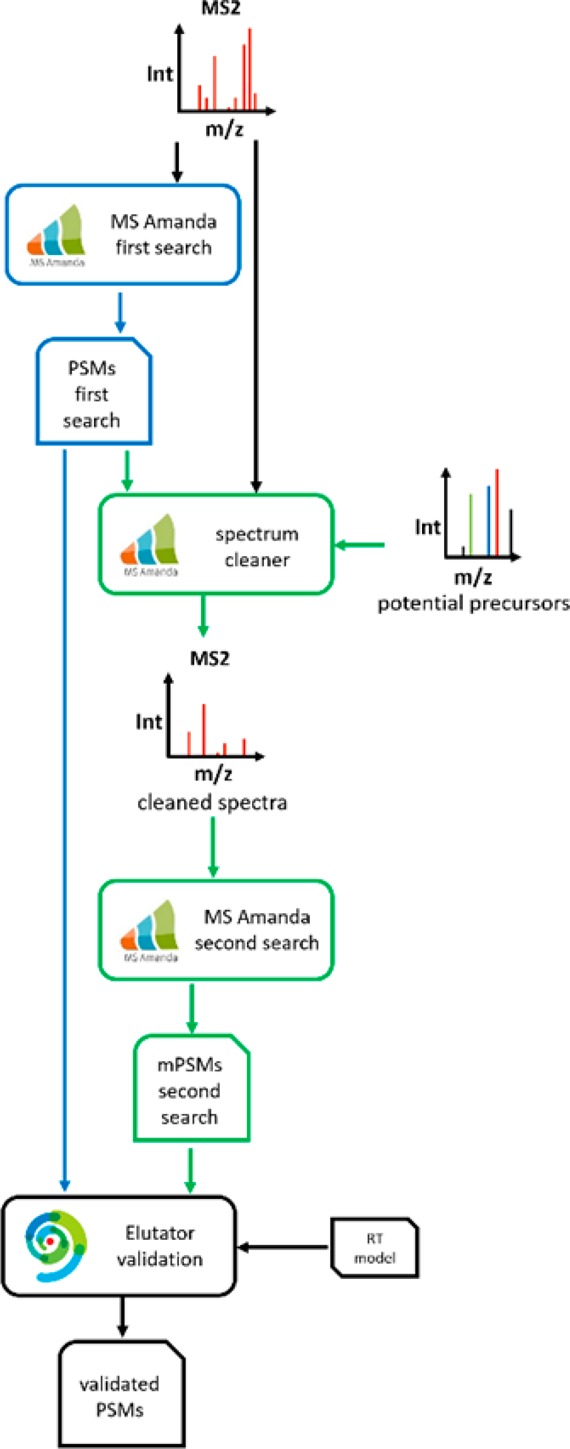
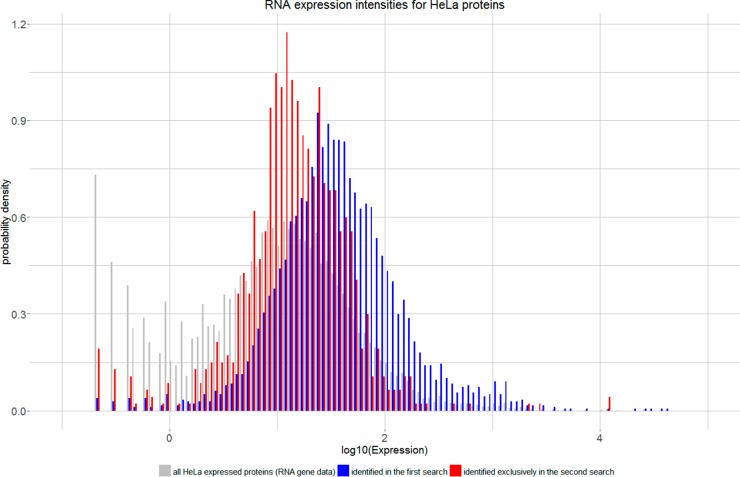
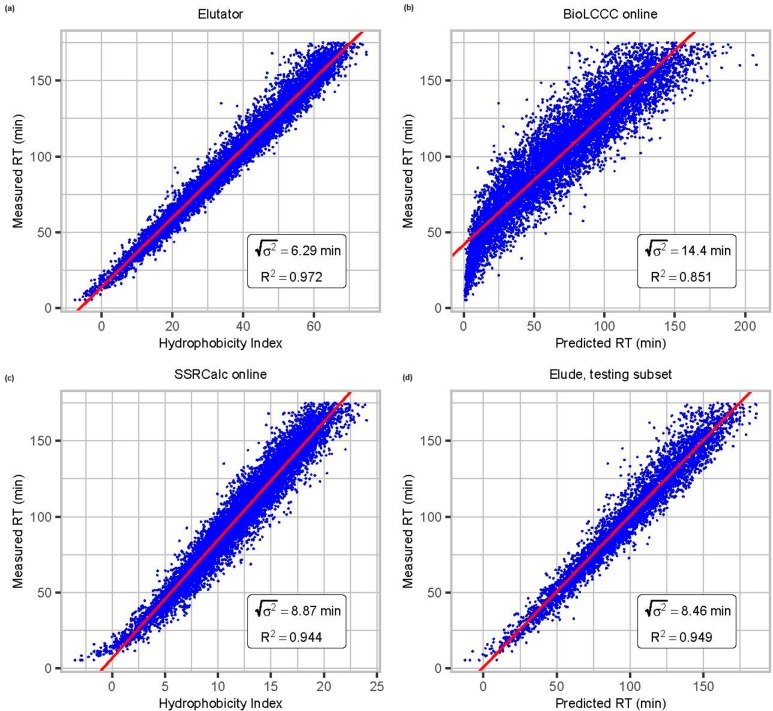
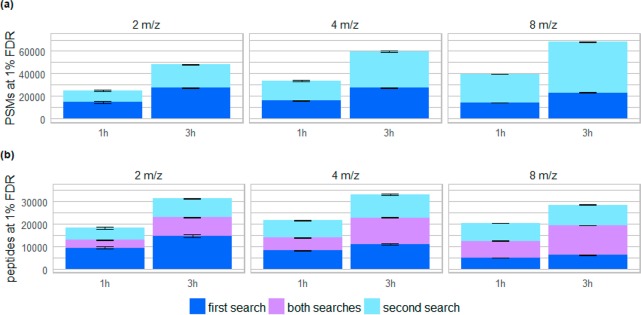
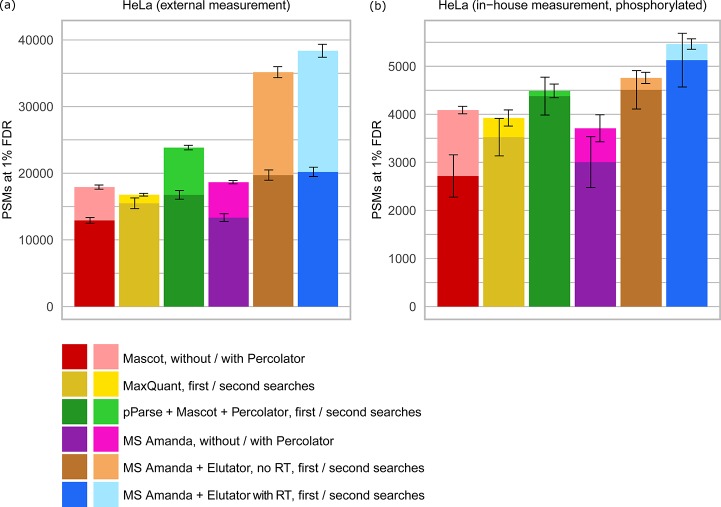
Coeluting peptides are still a major challenge for the identification and validation of MS/MS spectra, but carry great potential. To tackle these problems, we have developed the here presented CharmeRT workflow, combining a chimeric spectra identification strategy implemented as part of the MS Amanda algorithm with the validation system Elutator, which incorporates a highly accurate retention time prediction algorithm. For high-resolution data sets this workflow identifies 38-64% chimeric spectra, which results in up to 63% more unique peptides compared to a conventional single search strategy.
共洗脱肽仍然是鉴定和验证 MS/MS 谱的主要挑战,但具有巨大的潜力。为了解决这些问题,我们开发了这里提出的 CharmeRT 工作流程,该流程结合了作为 MS Amanda 算法一部分实现的嵌合谱识别策略和验证系统 Elutator,后者包含高度准确的保留时间预测算法。对于高分辨率数据集,该工作流程可识别 38%-64%的嵌合谱,与传统的单一搜索策略相比,可获得多达 63%的更多独特肽。