Evolutionary Genomics, Max Planck Institute for Evolutionary Biology, Plön, Germany.
Institute for Experimental Medicine, Kiel University, Kiel, Germany.
Microbiome. 2019 Sep 14;7(1):133. doi: 10.1186/s40168-019-0743-1.
The interplay between hosts and their associated microbiome is now recognized as a fundamental basis of the ecology, evolution, and development of both players. These interdependencies inspired a new view of multicellular organisms as "metaorganisms." The goal of the Collaborative Research Center "Origin and Function of Metaorganisms" is to understand why and how microbial communities form long-term associations with hosts from diverse taxonomic groups, ranging from sponges to humans in addition to plants.
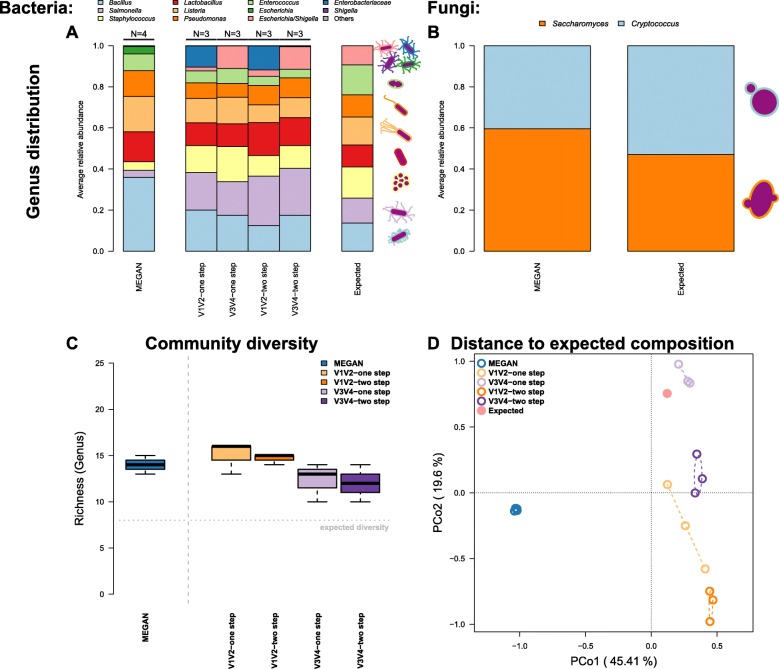
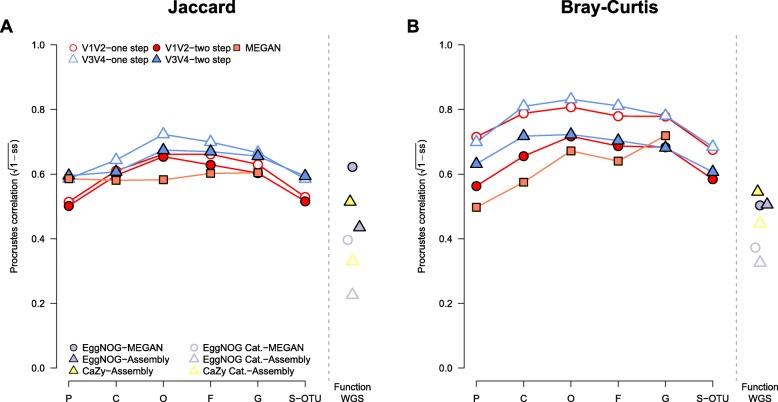
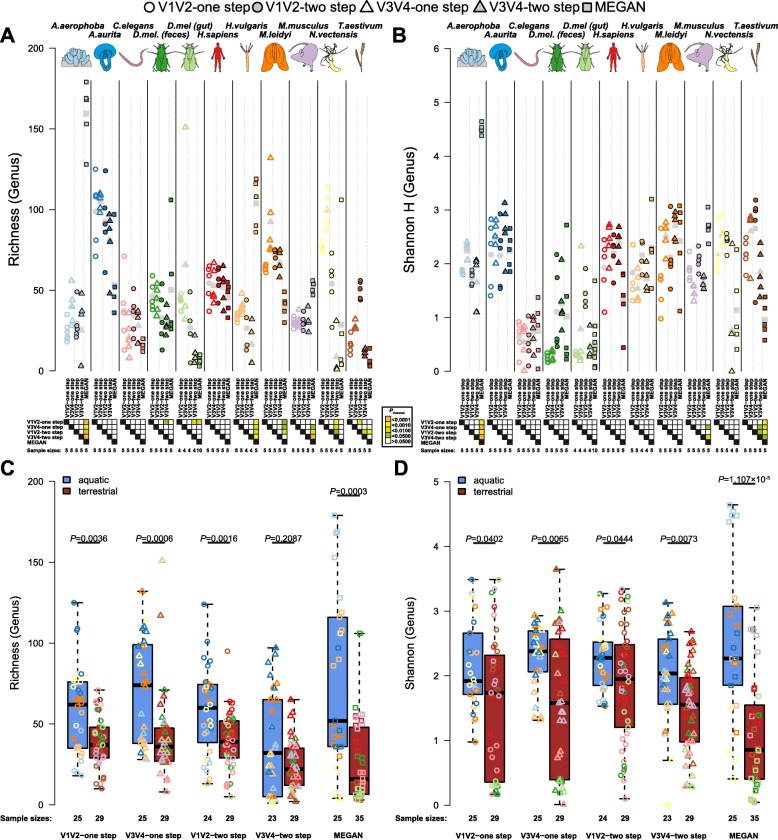
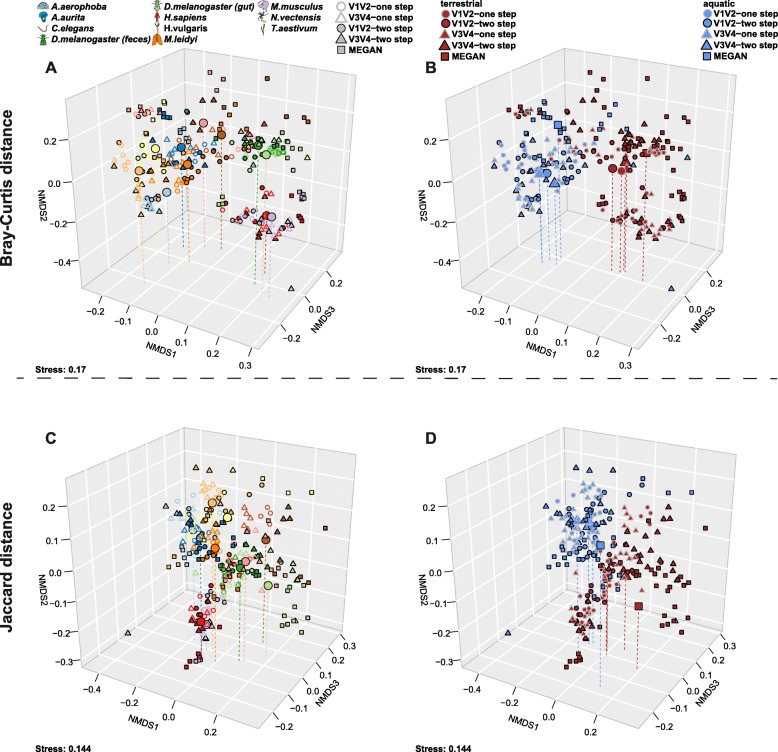
In order to optimize the choice of analysis procedures, which may differ according to the host organism and question at hand, we systematically compared the two main technical approaches for profiling microbial communities, 16S rRNA gene amplicon and metagenomic shotgun sequencing across our panel of ten host taxa. This includes two commonly used 16S rRNA gene regions and two amplification procedures, thus totaling five different microbial profiles per host sample.
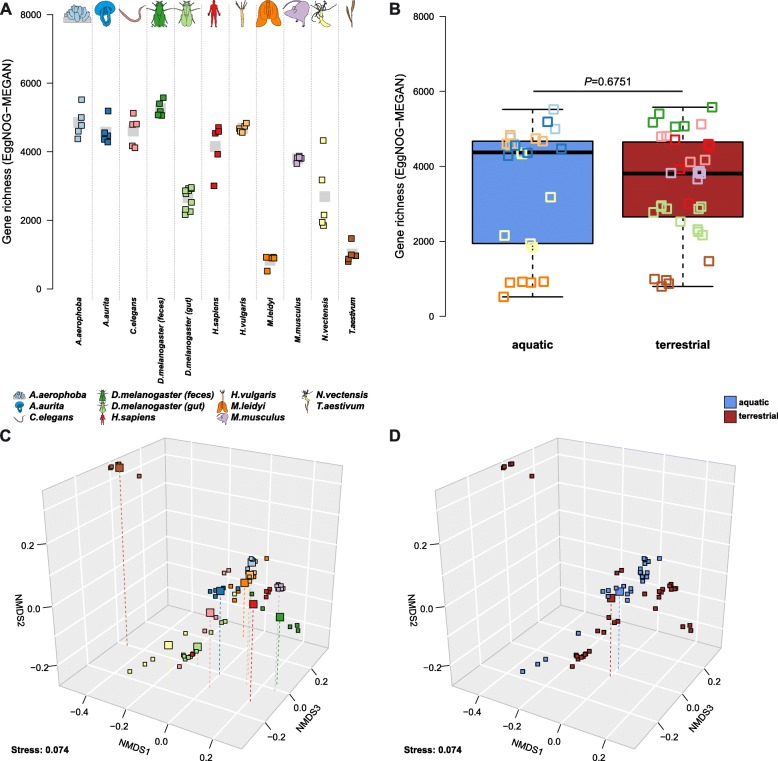
While 16S rRNA gene-based analyses are subject to much skepticism, we demonstrate that many aspects of bacterial community characterization are consistent across methods. The resulting insight facilitates the selection of appropriate methods across a wide range of host taxa. Overall, we recommend single- over multi-step amplification procedures, and although exceptions and trade-offs exist, the V3 V4 over the V1 V2 region of the 16S rRNA gene. Finally, by contrasting taxonomic and functional profiles and performing phylogenetic analysis, we provide important and novel insight into broad evolutionary patterns among metaorganisms, whereby the transition of animals from an aquatic to a terrestrial habitat marks a major event in the evolution of host-associated microbial composition.
宿主与其相关微生物组之间的相互作用现在被认为是两者生态、进化和发展的基础。这些相互依存关系激发了一种新的观点,即多细胞生物是“后生生物”。“后生生物的起源和功能”合作研究中心的目标是了解为什么以及微生物群落如何与来自不同分类群的宿主形成长期的联系,这些宿主的范围从海绵到人类以及植物。
为了优化分析程序的选择,这些程序可能因宿主生物和手头的问题而异,我们系统地比较了 profiling 微生物群落的两种主要技术方法,16S rRNA 基因扩增子和宏基因组鸟枪法测序,涵盖了我们的十个宿主分类群。这包括两个常用的 16S rRNA 基因区域和两种扩增程序,因此每个宿主样本有五个不同的微生物图谱。
虽然基于 16S rRNA 基因的分析受到了很多质疑,但我们证明了细菌群落特征的许多方面在方法上是一致的。由此产生的见解有助于在广泛的宿主分类群中选择合适的方法。总的来说,我们建议使用单步而非多步扩增程序,尽管存在例外和权衡,但 16S rRNA 基因的 V3-V4 区优于 V1-V2 区。最后,通过对比分类和功能图谱并进行系统发育分析,我们提供了后生生物中广泛的进化模式的重要和新的见解,其中动物从水生到陆生栖息地的转变标志着宿主相关微生物组成进化中的一个重大事件。