Meng Qingwei, Luo Zhang, Cao Chunyu, Sun Shishuai, Ma Qingquan, Li Zhongyu, Shi Baoming, Shan Anshan
Institute of Animal Nutrition, Northeast Agricultural University, Harbin, China.
Front Microbiol. 2020 Apr 17;11:694. doi: 10.3389/fmicb.2020.00694. eCollection 2020.
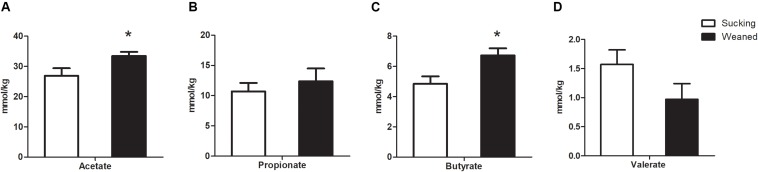
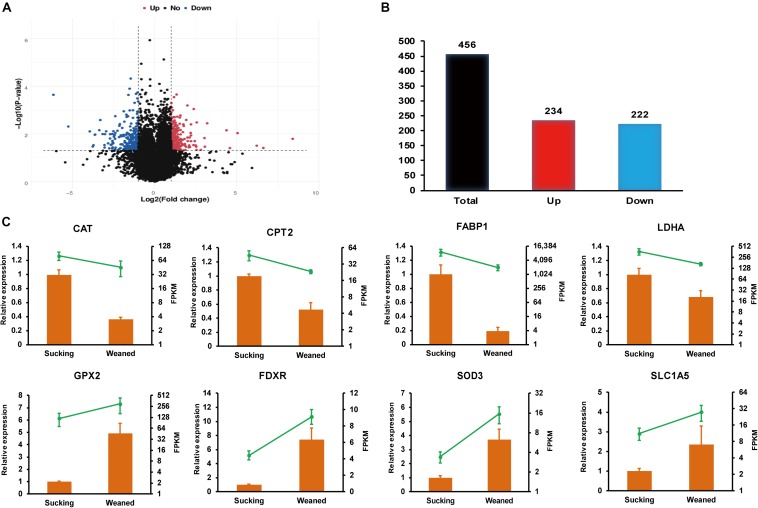
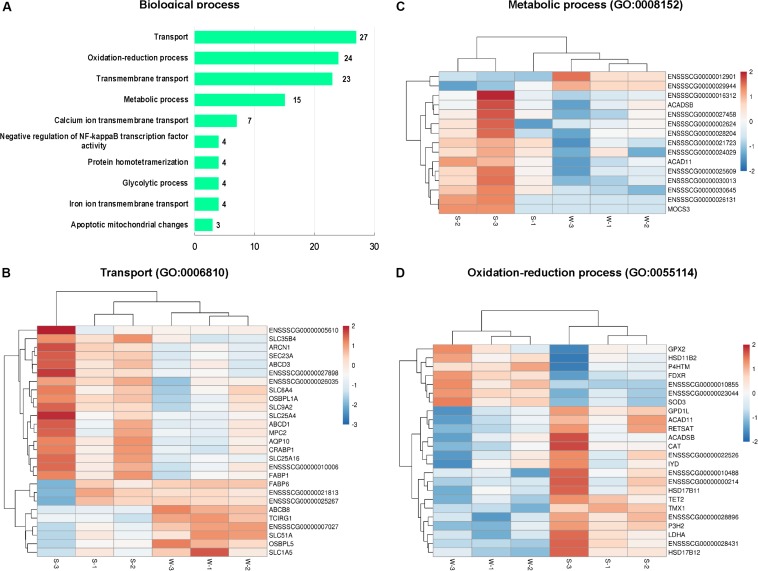
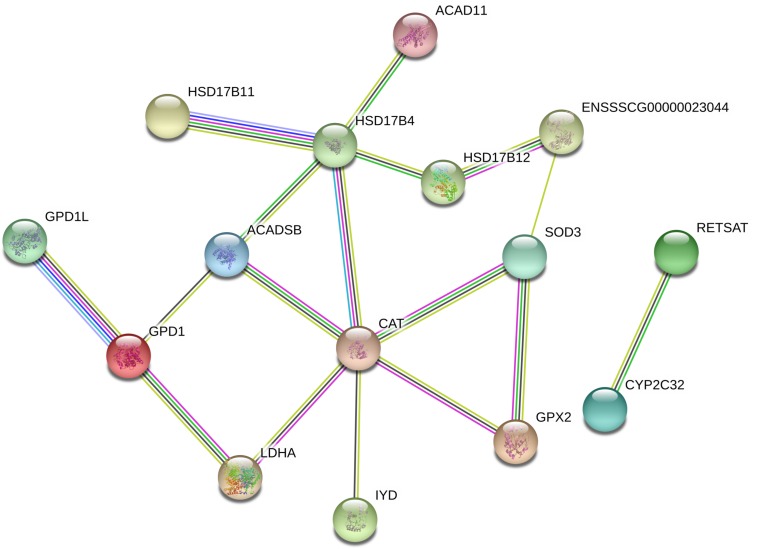
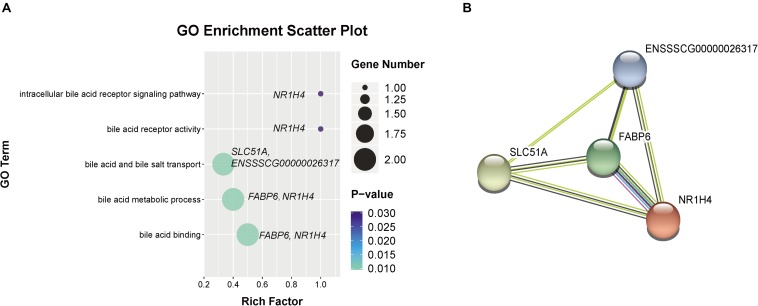
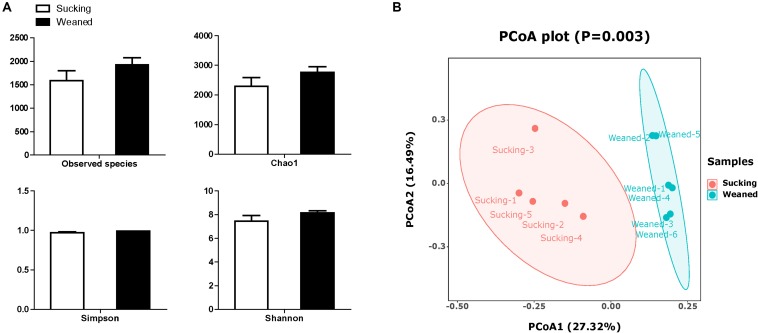
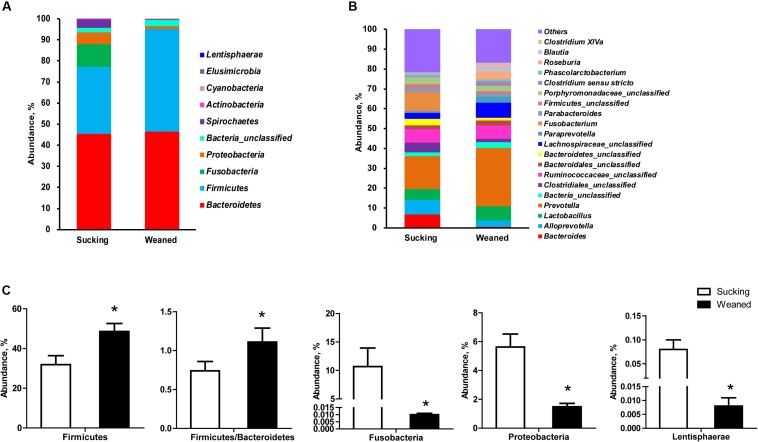
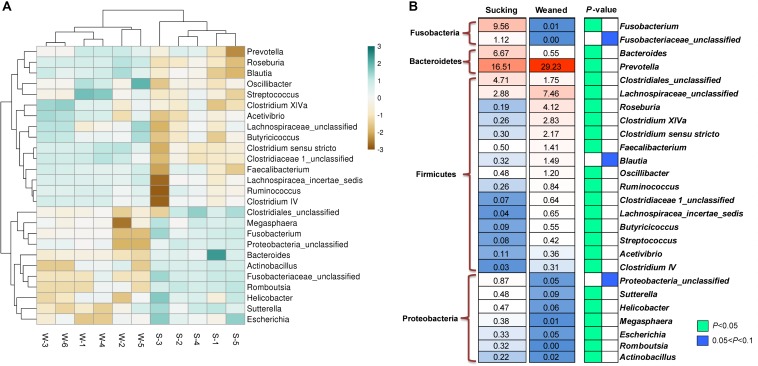
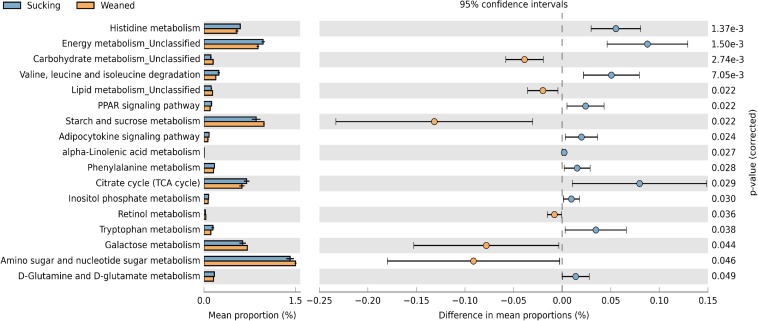
Weaning transition usually impairs intestinal architecture and functions and results in gut-associated disorders in pigs. Understanding the changes in intestinal transcriptome and gut microbiota during weaning transition is important for elucidating the underlying mechanism of weaning stress. In the present study, we performed RNA-seq to determine the changes in intestinal transcriptome and 16S rRNA sequencing to measure the gut microbiota changes in the weaning transition. Transcriptome results indicated that weaning transition altered intestinal gene expression involved in nutrient transport and metabolism. Regarding fatty metabolism, fatty acid-binding protein 1 (), acyl-CoA dehydrogenase (), and carnitine palmitoyltransferase 2 () expression in the intestine was decreased by weaning. Genes related to bile acid metabolism were increased by weaning, including , farnesoid X receptor ( or ) and organic solute transporter-α (). In addition, genes associated with oxidative stress were altered by weaning transition, including decreased catalase () and lactate dehydrogenase () and increased glutathione peroxidase 2 () and superoxide dismutase 3 (). Results of microbiota composition showed that the Firmicutes abundance and Firmicutes/Bacteroidetes ratio were increased and that the Proteobacteria abundance in the fecal microbiota was decreased by the weaning process; during the weaning transition, the and abundances decreased markedly, and these bacteria nearly disappeared, while the abundance showed a marked increase. Moreover, the levels of the microbial metabolites butyrate and acetate increased with changes in gut microbiota composition. In addition, predictive metagenome by PICRUSt analysis showed that the pathways related to D-glutamine and D-glutamate metabolism, citrate cycle (TCA cycle), peroxisome proliferators-activated receptor (PPAR) signaling, alpha-linolenic acid metabolism were decreased and the pathway related to retinol metabolism was increased in the gut microbiota of piglets during weaning transition. Our results showed that early weaning alters intestinal gene expression involved in nutrient metabolism, which may be due to the changes in microbiota composition.
断奶过渡通常会损害猪的肠道结构和功能,并导致肠道相关疾病。了解断奶过渡期间肠道转录组和肠道微生物群的变化对于阐明断奶应激的潜在机制很重要。在本研究中,我们进行了RNA测序以确定肠道转录组的变化,并进行16S rRNA测序以测量断奶过渡期间肠道微生物群的变化。转录组结果表明,断奶过渡改变了参与营养物质运输和代谢的肠道基因表达。在脂肪代谢方面,断奶使肠道中脂肪酸结合蛋白1(FABP1)、酰基辅酶A脱氢酶(ACAD)和肉碱棕榈酰转移酶2(CPT2)的表达降低。与胆汁酸代谢相关的基因在断奶后增加,包括CYP7A1、法尼酯X受体(FXR或NR1H4)和有机溶质转运蛋白-α(OSTα)。此外,与氧化应激相关的基因在断奶过渡时发生改变,包括过氧化氢酶(CAT)和乳酸脱氢酶(LDH)降低,而谷胱甘肽过氧化物酶2(GPX2)和超氧化物歧化酶3(SOD3)增加。微生物群组成结果显示,断奶过程使粪便微生物群中厚壁菌门丰度和厚壁菌门/拟杆菌门比率增加,而变形菌门丰度降低;在断奶过渡期间,梭菌属和双歧杆菌属丰度显著下降,这些细菌几乎消失,而肠杆菌属丰度显著增加。此外,微生物代谢产物丁酸和乙酸的水平随着肠道微生物群组成的变化而增加。此外,通过PICRUSt分析的预测宏基因组显示,在断奶过渡期间,仔猪肠道微生物群中与D-谷氨酰胺和D-谷氨酸代谢、柠檬酸循环(TCA循环)、过氧化物酶体增殖物激活受体(PPAR)信号传导、α-亚麻酸代谢相关的途径减少,而与视黄醇代谢相关的途径增加。我们的结果表明,早期断奶改变了参与营养代谢的肠道基因表达,这可能是由于微生物群组成的变化所致。