Hossain Md Delowar, Huang Yufeng, Yu Ted H, Goddard William A, Luo Zhengtang
Department of Chemical and Biological Engineering, William Mong Institute of Nano Science and Technology, and Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong.
Materials and Process Simulation Center (mc 134-74), California Institute of Technology, Pasadena, CA, 91125, USA.
Nat Commun. 2020 May 7;11(1):2256. doi: 10.1038/s41467-020-16119-6.
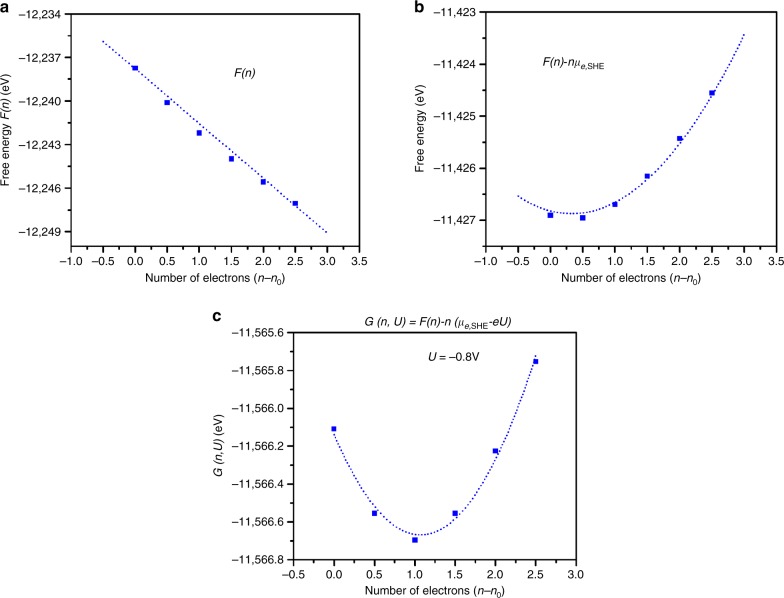
Experiments have shown that graphene-supported Ni-single atom catalysts (Ni-SACs) provide a promising strategy for the electrochemical reduction of CO to CO, but the nature of the Ni sites (Ni-NC, Ni-NC, Ni-N) in Ni-SACs has not been determined experimentally. Here, we apply the recently developed grand canonical potential kinetics (GCP-K) formulation of quantum mechanics to predict the kinetics as a function of applied potential (U) to determine faradic efficiency, turn over frequency, and Tafel slope for CO and H production for all three sites. We predict an onset potential (at 10 mA cm) U = -0.84 V (vs. RHE) for Ni-NC site and U = -0.92 V for Ni-NC site in agreement with experiments, and U = -1.03 V for Ni-N. We predict that the highest current is for Ni-N, leading to 700 mA cm at U = -1.12 V. To help determine the actual sites in the experiments, we predict the XPS binding energy shift and CO vibrational frequency for each site.
实验表明,石墨烯负载的镍单原子催化剂(Ni-SACs)为将CO电化学还原为CO提供了一种有前景的策略,但Ni-SACs中镍位点(Ni-NC、Ni-NC、Ni-N)的性质尚未通过实验确定。在此,我们应用最近开发的量子力学巨正则势动力学(GCP-K)公式来预测动力学与外加电势(U)的函数关系,以确定所有三个位点产生CO和H的法拉第效率、周转频率和塔菲尔斜率。我们预测Ni-NC位点在10 mA cm时的起始电势U = -0.84 V(相对于可逆氢电极),Ni-NC位点为U = -0.92 V,与实验结果一致,Ni-N位点为U = -1.03 V。我们预测Ni-N的电流最高,在U = -1.12 V时可达700 mA cm。为了帮助确定实验中的实际位点,我们预测了每个位点的XPS结合能位移和CO振动频率。