Center for Research and Formation in Artificial Intelligence, Universidad de los Andes, Bogotá, Colombia.
Department of Biomedical Engineering, Universidad de los Andes, Bogotá, Colombia.
PLoS One. 2021 Apr 26;16(4):e0241728. doi: 10.1371/journal.pone.0241728. eCollection 2021.
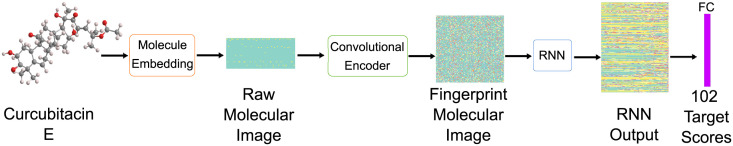
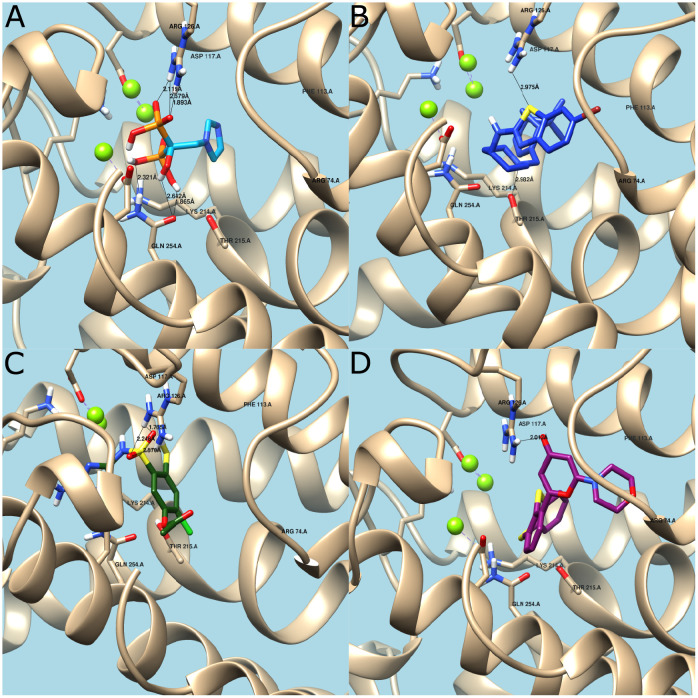
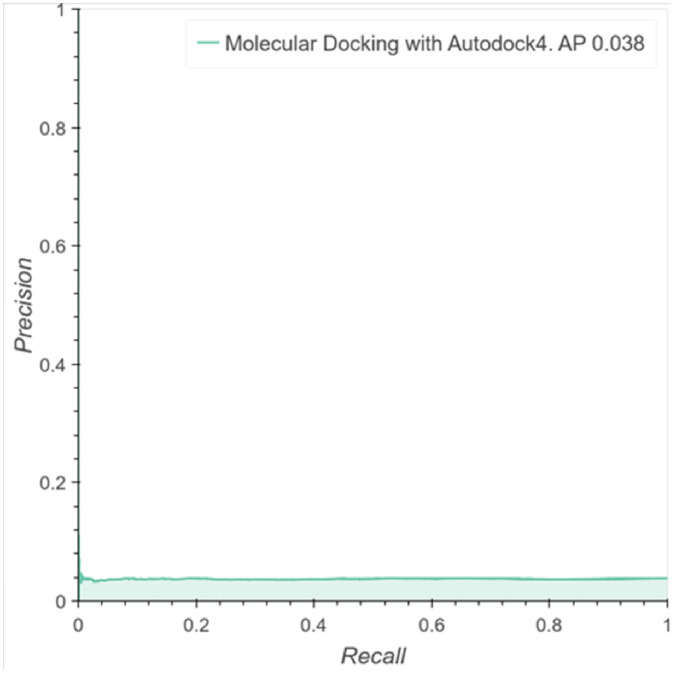
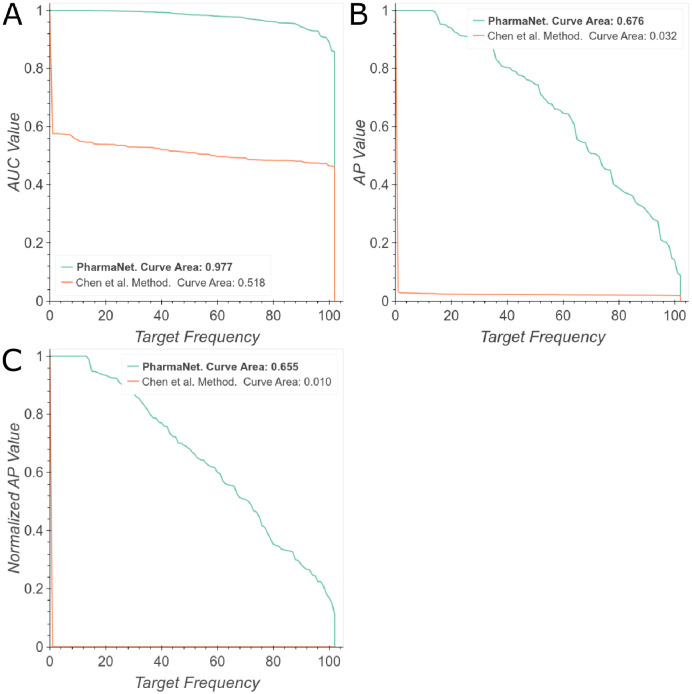
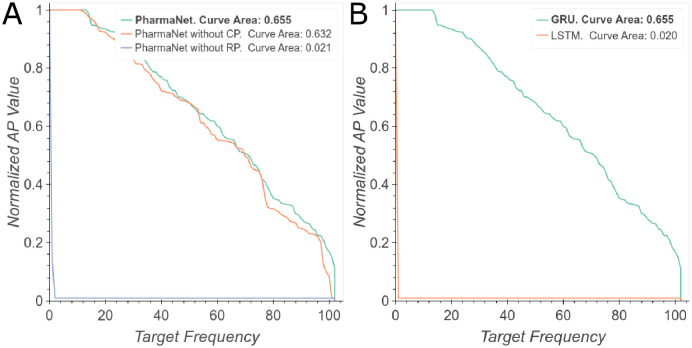
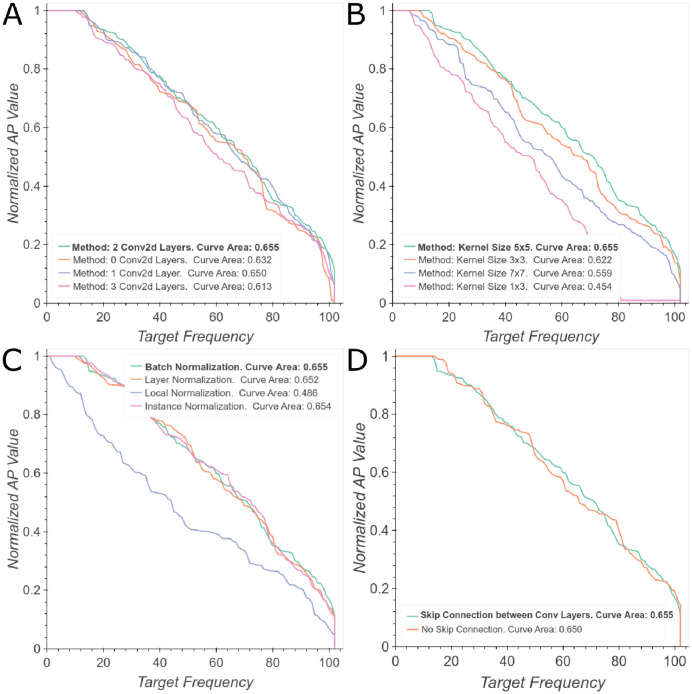
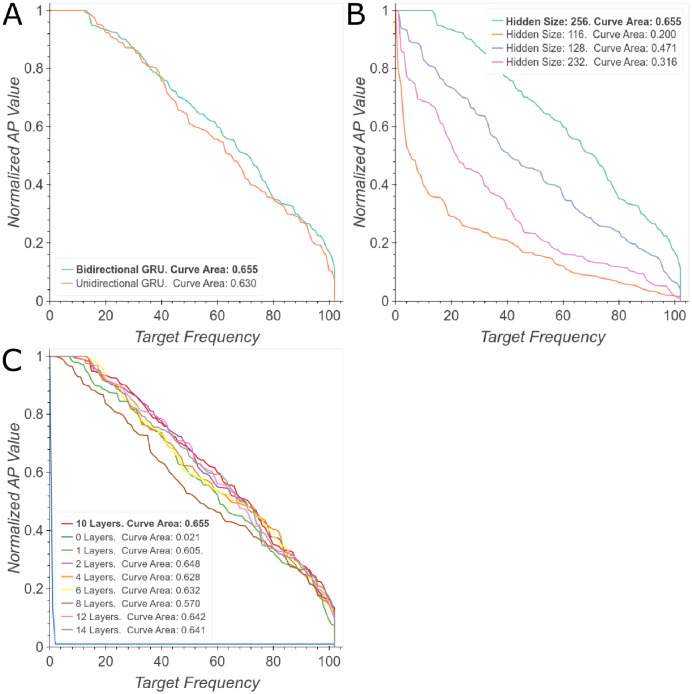
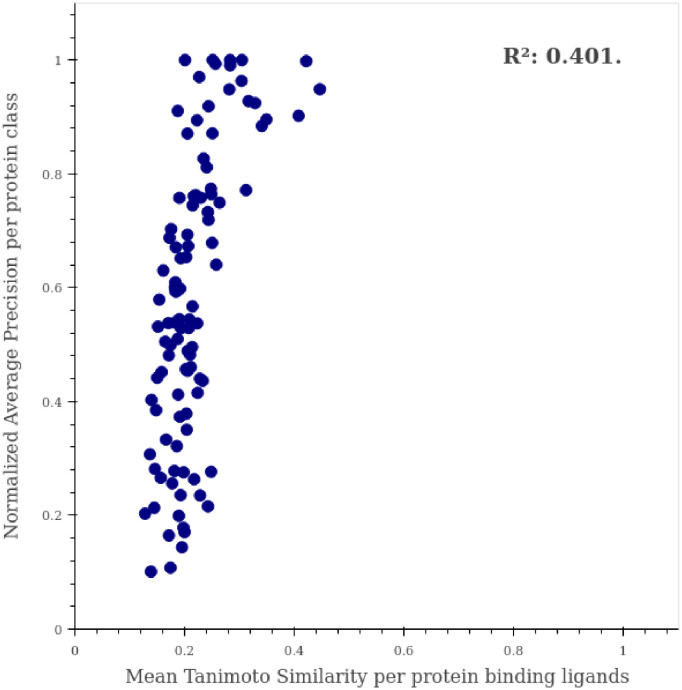
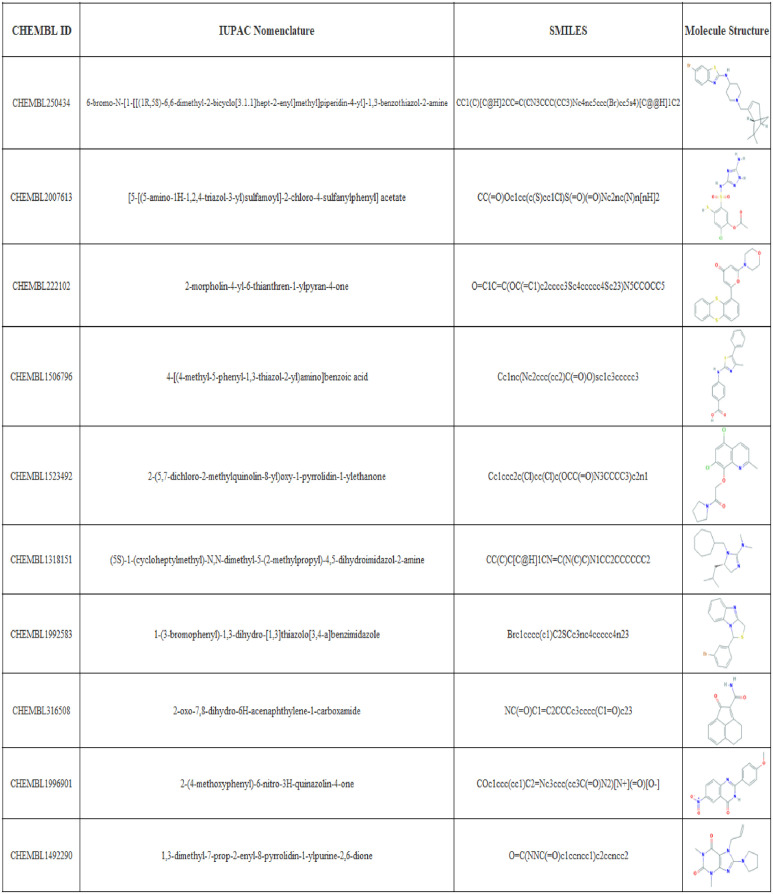
The discovery and development of novel pharmaceuticals is an area of active research mainly due to the large investments required and long payback times. As of 2016, the development of a novel drug candidate required up to $ USD 2.6 billion in investment for only 10% rate of approval by the FDA. To help decreasing the costs associated with the process, a number of in silico approaches have been developed with relatively low success due to limited predicting performance. Here, we introduced a machine learning-based algorithm as an alternative for a more accurate search of new pharmacological candidates, which takes advantage of Recurrent Neural Networks (RNN) for active molecule prediction within large databases. Our approach, termed PharmaNet was implemented here to search for ligands against specific cell receptors within 102 targets of the DUD-E database, which contains 22886 active molecules. PharmaNet comprises three main phases. First, a SMILES representation of the molecule is converted into a raw molecular image. Second, a convolutional encoder processes the data to obtain a fingerprint molecular image that is finally analyzed by a Recurrent Neural Network (RNN). This approach enables precise predictions of the molecules' target on the basis of the feature extraction, the sequence analysis and the relevant information filtered out throughout the process. Molecule Target prediction is a highly unbalanced detection problem and therefore, we propose that an adequate evaluation metric of performance is the area under the Normalized Average Precision (NAP) curve. PharmaNet largely surpasses the previous state-of-the-art method with 97.7% in the Receiver Operating Characteristic curve (ROC-AUC) and 65.5% in the NAP curve. We obtained a perfect performance for human farnesyl pyrophosphate synthase (FPPS), which is a potential target for antimicrobial and anticancer treatments. We decided to test PharmaNet for activity prediction against FPPS by searching in the CHEMBL data set. We obtained three (3) potential inhibitors that were further validated through both molecular docking and in silico toxicity prediction. Most importantly, one of this candidates, CHEMBL2007613, was predicted as a potential antiviral due to its involvement on the PCDH17 pathway, which has been reported to be related to viral infections.
新型药物的发现和开发是一个活跃的研究领域,主要是因为需要大量投资和较长的投资回报期。截至 2016 年,开发一种新的药物候选物需要高达 26 亿美元的投资,而只有 10%的药物能通过美国食品和药物管理局(FDA)的批准。为了帮助降低与该过程相关的成本,已经开发了许多基于计算机的方法,但由于预测性能有限,相对成功率较低。在这里,我们引入了一种基于机器学习的算法,作为更准确地搜索新的药理候选物的替代方法,该方法利用递归神经网络(RNN)在大型数据库中对活性分子进行预测。我们的方法称为 PharmaNet,用于在 DUD-E 数据库的 102 个靶点中搜索针对特定细胞受体的配体,该数据库包含 22886 个活性分子。PharmaNet 由三个主要阶段组成。首先,将分子的 SMILES 表示形式转换为原始分子图像。其次,卷积编码器处理数据以获得指纹分子图像,最后由递归神经网络(RNN)对其进行分析。该方法通过特征提取、序列分析和整个过程中过滤出的相关信息,能够精确地预测分子的靶点。分子靶点预测是一个高度不平衡的检测问题,因此,我们建议使用归一化平均精度(NAP)曲线下的面积作为性能的适当评估指标。PharmaNet 在接收器工作特征曲线(ROC-AUC)中以 97.7%和 NAP 曲线中以 65.5%的优势大大超过了以前的最先进方法。我们在人法呢基二磷酸合酶(FPPS)上获得了完美的性能,FPPS 是抗菌和抗癌治疗的潜在靶点。我们决定通过在 CHEMBL 数据集上搜索来测试 PharmaNet 对 FPPS 的活性预测。我们得到了三个(3)种潜在的抑制剂,通过分子对接和计算机毒性预测进一步验证。最重要的是,候选药物之一 CHEMBL2007613 被预测为一种潜在的抗病毒药物,因为它参与了 PCDH17 途径,该途径已被报道与病毒感染有关。