Hsing Michael, Byler Kendall Grant, Cherkasov Artem
Faculty of Graduate Studies, Bioinformatics Graduate Program, University of British Columbia, Vancouver, BC, Canada.
BMC Syst Biol. 2008 Sep 16;2:80. doi: 10.1186/1752-0509-2-80.
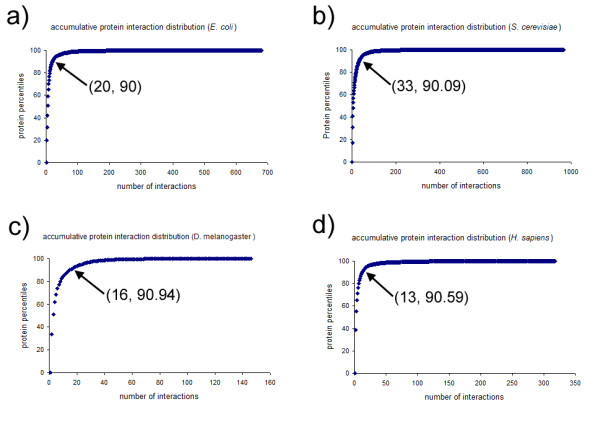
Protein-protein interactions mediate a wide range of cellular functions and responses and have been studied rigorously through recent large-scale proteomics experiments and bioinformatics analyses. One of the most important findings of those endeavours was the observation that 'hub' proteins participate in significant numbers of protein interactions and play critical roles in the organization and function of cellular protein interaction networks (PINs) 12. It has also been demonstrated that such hub proteins may constitute an important pool of attractive drug targets.Thus, it is crucial to be able to identify hub proteins based not only on experimental data but also by means of bioinformatics predictions.
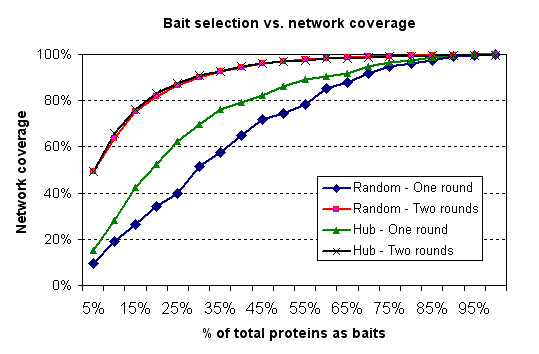
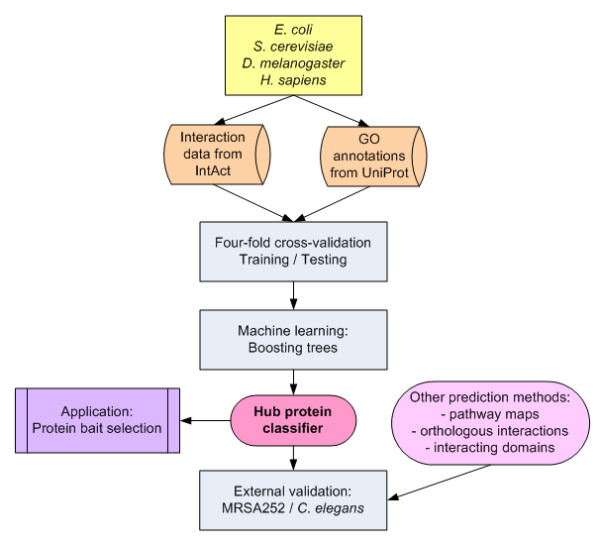
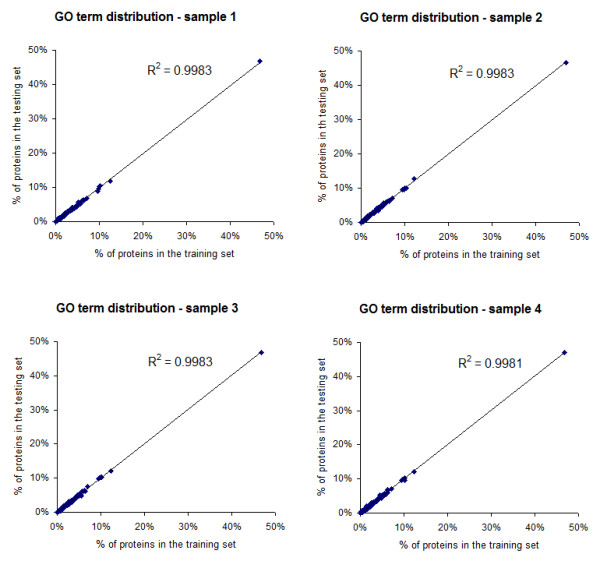
A hub protein classifier has been developed based on the available interaction data and Gene Ontology (GO) annotations for proteins in the Escherichia coli, Saccharomyces cerevisiae, Drosophila melanogaster and Homo sapiens genomes. In particular, by utilizing the machine learning method of boosting trees we were able to create a predictive bioinformatics tool for the identification of proteins that are likely to play the role of a hub in protein interaction networks. Testing the developed hub classifier on external sets of experimental protein interaction data in Methicillin-resistant Staphylococcus aureus (MRSA) 252 and Caenorhabditis elegans demonstrated that our approach can predict hub proteins with a high degree of accuracy.A practical application of the developed bioinformatics method has been illustrated by the effective protein bait selection for large-scale pull-down experiments that aim to map complete protein-protein interaction networks for several species.
The successful development of an accurate hub classifier demonstrated that highly-connected proteins tend to share certain relevant functional properties reflected in their Gene Ontology annotations. It is anticipated that the developed bioinformatics hub classifier will represent a useful tool for the theoretical prediction of highly-interacting proteins, the study of cellular network organizations, and the identification of prospective drug targets - even in those organisms that currently lack large-scale protein interaction data.
蛋白质-蛋白质相互作用介导了广泛的细胞功能和反应,并且通过近期的大规模蛋白质组学实验和生物信息学分析得到了深入研究。这些研究中最重要的发现之一是观察到“枢纽”蛋白参与大量的蛋白质相互作用,并在细胞蛋白质相互作用网络(PINs)的组织和功能中发挥关键作用。研究还表明,这类枢纽蛋白可能构成了有吸引力的药物靶点的重要来源。因此,不仅能够基于实验数据,还能借助生物信息学预测来识别枢纽蛋白至关重要。
基于大肠杆菌、酿酒酵母、黑腹果蝇和人类基因组中蛋白质的可用相互作用数据和基因本体(GO)注释,开发了一种枢纽蛋白分类器。特别是,通过利用提升树的机器学习方法,我们能够创建一种预测性生物信息学工具,用于识别可能在蛋白质相互作用网络中发挥枢纽作用的蛋白质。在耐甲氧西林金黄色葡萄球菌(MRSA)252和秀丽隐杆线虫的外部实验性蛋白质相互作用数据集上测试所开发的枢纽分类器,结果表明我们的方法能够高度准确地预测枢纽蛋白。通过为旨在绘制几个物种完整蛋白质-蛋白质相互作用网络的大规模下拉实验有效选择蛋白质诱饵,说明了所开发的生物信息学方法的实际应用。
准确的枢纽分类器的成功开发表明,高度连接的蛋白质往往共享其基因本体注释中反映的某些相关功能特性。预计所开发的生物信息学枢纽分类器将成为用于理论预测高度相互作用蛋白质、研究细胞网络组织以及识别潜在药物靶点的有用工具——即使在目前缺乏大规模蛋白质相互作用数据的生物体中也是如此。