Janssen Stefan, McDonald Daniel, Gonzalez Antonio, Navas-Molina Jose A, Jiang Lingjing, Xu Zhenjiang Zech, Winker Kevin, Kado Deborah M, Orwoll Eric, Manary Mark, Mirarab Siavash, Knight Rob
Department of Pediatrics, University of California San Diego, La Jolla, California, USA.
Department of Computer Science and Engineering, University of California San Diego, La Jolla, California, USA.
mSystems. 2018 Apr 17;3(3). doi: 10.1128/mSystems.00021-18. eCollection 2018 May-Jun.
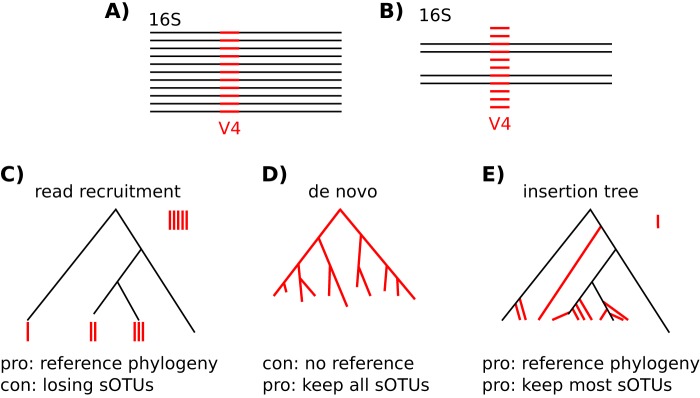
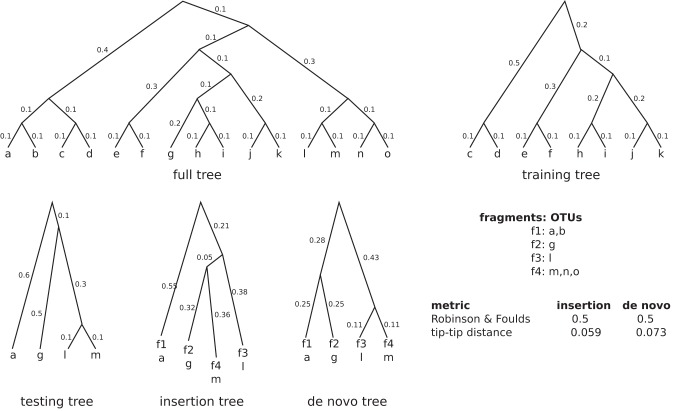
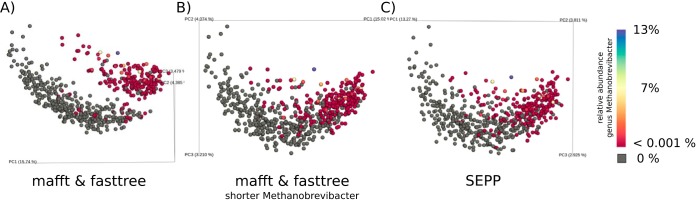
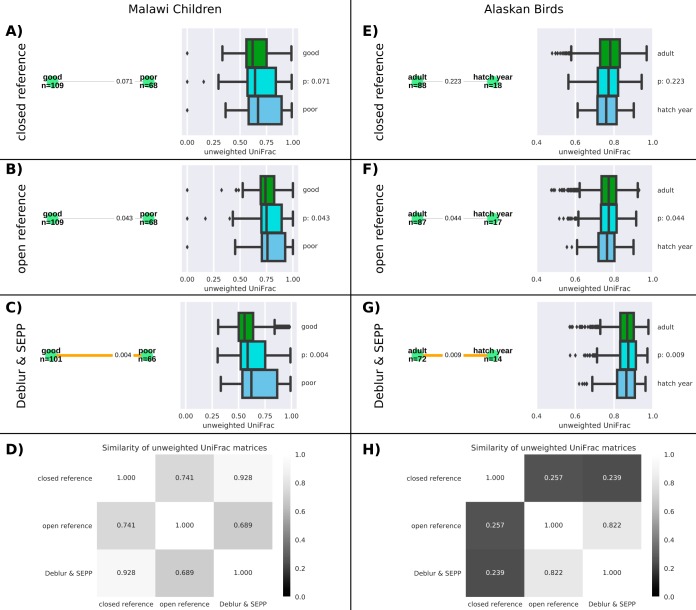
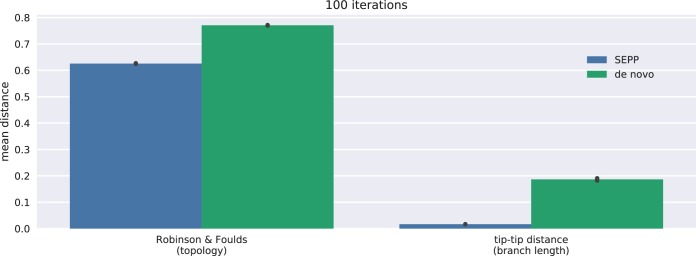
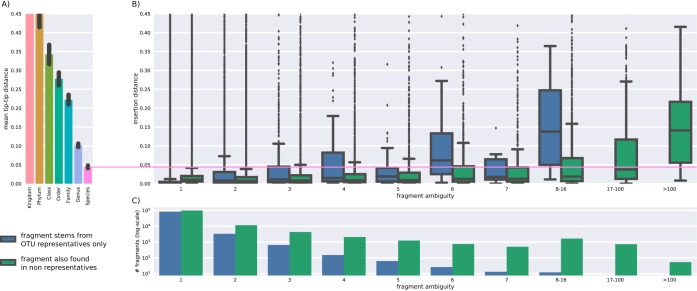
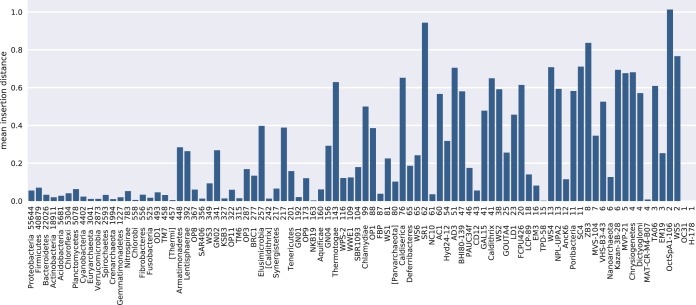
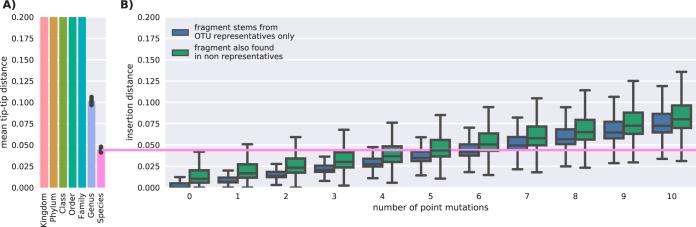
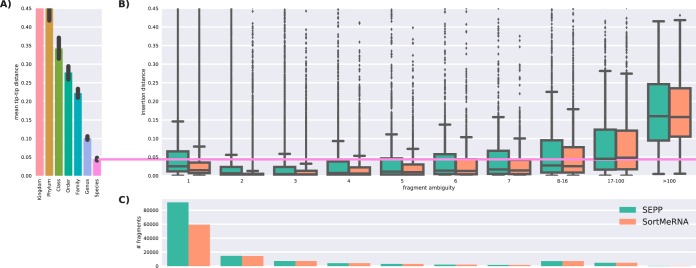
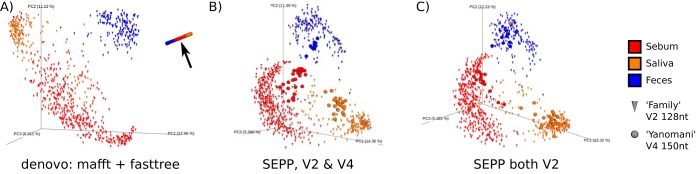
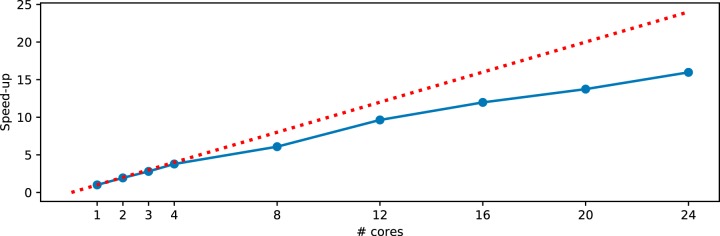
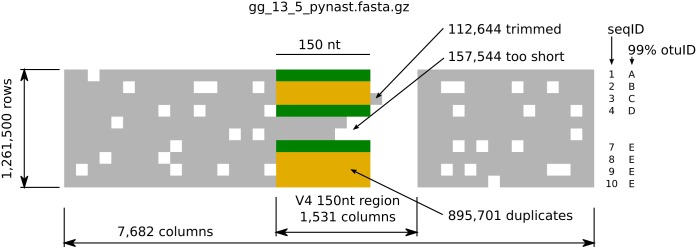
Recent algorithmic advances in amplicon-based microbiome studies enable the inference of exact amplicon sequence fragments. These new methods enable the investigation of sub-operational taxonomic units (sOTU) by removing erroneous sequences. However, short (e.g., 150-nucleotide [nt]) DNA sequence fragments do not contain sufficient phylogenetic signal to reproduce a reasonable tree, introducing a barrier in the utilization of critical phylogenetically aware metrics such as Faith's PD or UniFrac. Although fragment insertion methods do exist, those methods have not been tested for sOTUs from high-throughput amplicon studies in insertions against a broad reference phylogeny. We benchmarked the SATé-enabled phylogenetic placement (SEPP) technique explicitly against 16S V4 sequence fragments and showed that it outperforms the conceptually problematic but often-used practice of reconstructing phylogenies. In addition, we provide a BSD-licensed QIIME2 plugin (https://github.com/biocore/q2-fragment-insertion) for SEPP and integration into the microbial study management platform QIITA. The move from OTU-based to sOTU-based analysis, while providing additional resolution, also introduces computational challenges. We demonstrate that one popular method of dealing with sOTUs (building a tree from the short sequences) can provide incorrect results in human gut metagenomic studies and show that phylogenetic placement of the new sequences with SEPP resolves this problem while also yielding other benefits over existing methods.
基于扩增子的微生物组研究中,近期算法的进展使得能够推断出确切的扩增子序列片段。这些新方法通过去除错误序列,能够对亚操作分类单元(sOTU)进行研究。然而,短的(例如150个核苷酸[nt])DNA序列片段不包含足够的系统发育信号来重建合理的树,这在使用诸如费思系统发育多样性(Faith's PD)或非加权 UniFrac 等关键的系统发育感知指标时形成了障碍。虽然存在片段插入方法,但这些方法尚未针对高通量扩增子研究中的sOTU在插入到广泛的参考系统发育树时进行测试。我们明确地将启用SATé的系统发育定位(SEPP)技术与16S V4序列片段进行了基准测试,结果表明它优于概念上有问题但经常使用的系统发育重建方法。此外,我们为SEPP提供了一个遵循BSD许可的QIIME2插件(https://github.com/biocore/q2-fragment-insertion),并将其集成到微生物研究管理平台QIITA中。从基于操作分类单元(OTU)的分析转向基于sOTU的分析,虽然提供了额外的分辨率,但也带来了计算挑战。我们证明,一种处理sOTU的常用方法(从短序列构建树)在人类肠道宏基因组研究中可能会产生错误结果,并表明使用SEPP对新序列进行系统发育定位可以解决这个问题,同时相对于现有方法还有其他优势。