Department of Theoretical Chemistry, Chemical Centre, Lund University, P. O. Box 124, 221 00, Lund, Sweden.
J Biol Inorg Chem. 2020 May;25(3):521-540. doi: 10.1007/s00775-020-01780-5. Epub 2020 Apr 7.
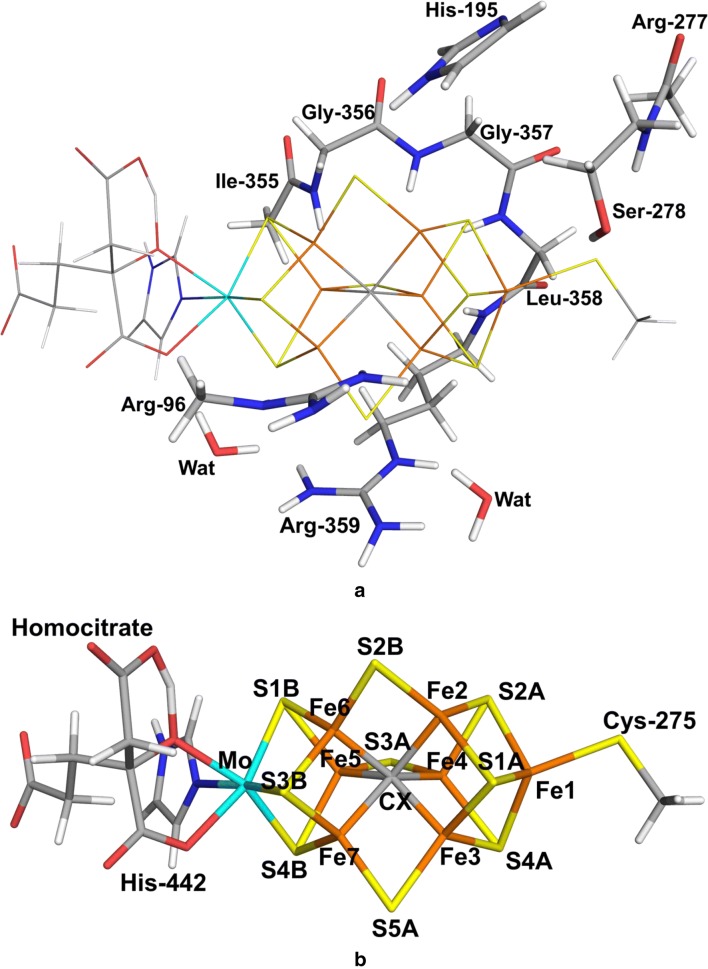
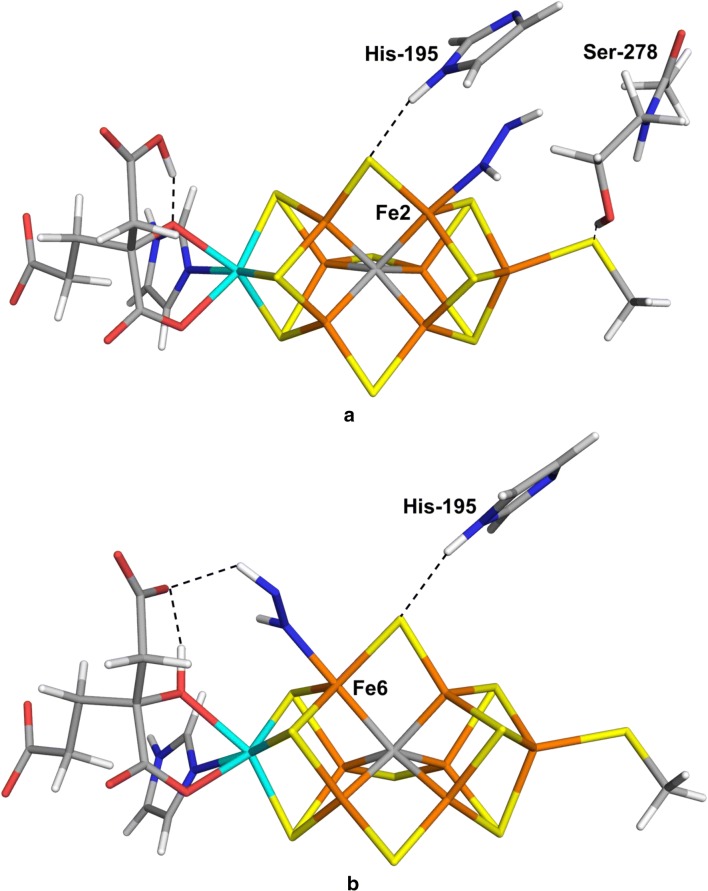
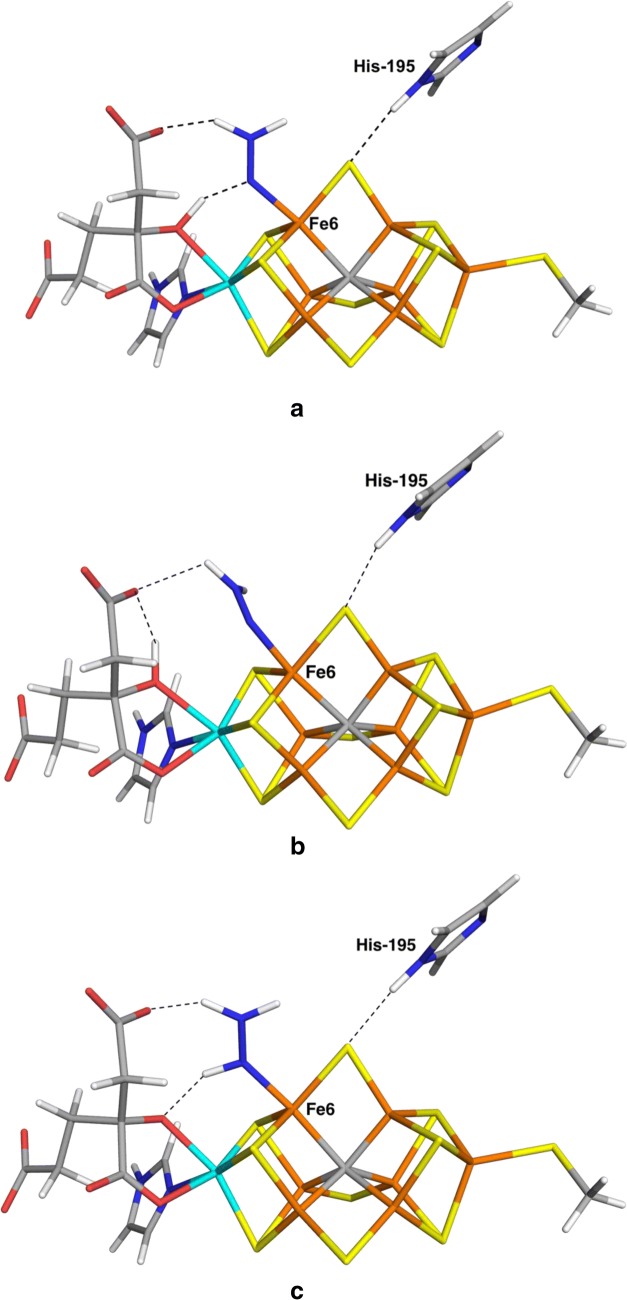
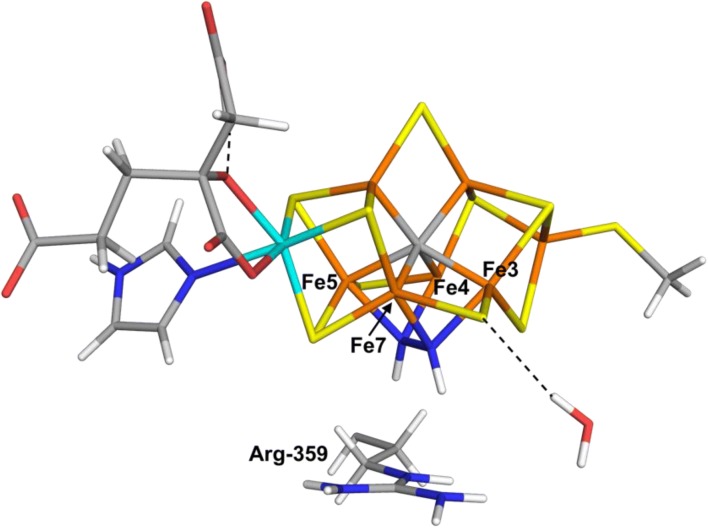
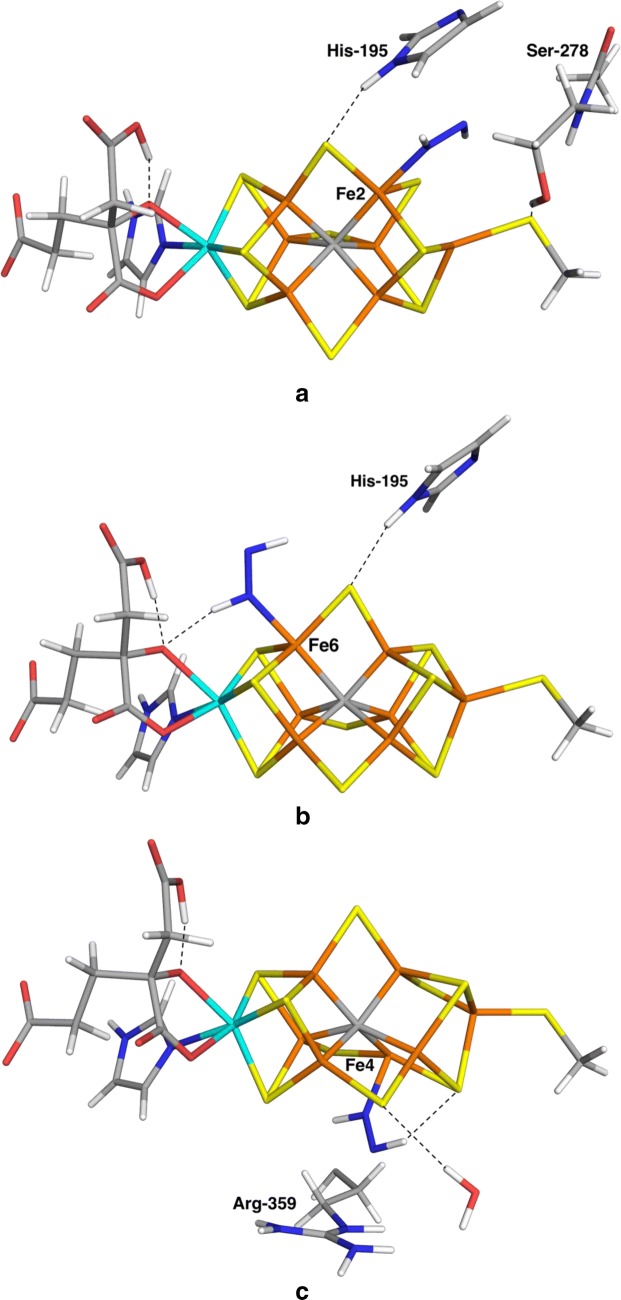
We have made a systematic combined quantum mechanical and molecular mechanical (QM/MM) investigation of possible structures of the N bound state of nitrogenase. We assume that N is immediately protonated to a NH state, thereby avoiding the problem of determining the position of the protons in the cluster. We have systematically studied both end-on and side-on structures, as well as both HNNH and NNH states. Our results indicate that the binding of NH is determined more by interactions and steric clashes with the surrounding protein than by the intrinsic preferences of the ligand and the cluster. The best binding mode with both the TPSS and B3LYP density-functional theory methods has trans-HNNH terminally bound to Fe2. It is stabilised by stacking of the substrate with His-195 and Ser-278. However, several other structures come rather close in energy (within 3-35 kJ/mol) at least in some calculations: The corresponding cis-HNNH structure terminally bound to Fe2 is second best with B3LYP. A structure with HNNH terminally bound to Fe6 is second most stable with TPSS (where the third proton is transferred to the substrate from the homocitrate ligand). Structures with trans-HNNH, bound to Fe4 or Fe6, or cis-HNNH bound to Fe6 are also rather stable. Finally, with the TPSS functional, a structure with cis-HNNH side-on binding to the Fe3-Fe4-Fe5-Fe7 face of the cluster is also rather low in energy, but all side-on structures are strongly disfavoured by the B3LYP method.
我们对固氮酶中 N 结合态的可能结构进行了系统的量子力学和分子力学(QM/MM)联合研究。我们假设 N 立即质子化为 NH 态,从而避免了在簇中确定质子位置的问题。我们系统地研究了端到端和侧到侧结构,以及 HNNH 和 NNH 态。我们的结果表明,NH 的结合更多地取决于与周围蛋白质的相互作用和空间位阻,而不是配体和簇的固有偏好。TPSS 和 B3LYP 密度泛函理论方法的最佳结合模式均具有末端与 Fe2 结合的反式 HNNH。它通过与 His-195 和 Ser-278 的底物堆积而稳定。然而,在某些计算中,其他几个结构的能量非常接近(在 3-35 kJ/mol 范围内):与 B3LYP 相比,末端与 Fe2 结合的相应顺式 HNNH 结构是第二好的。末端与 Fe6 结合的 HNNH 结构在 TPSS 中是第三稳定的(其中第三个质子从同型柠檬酸配体转移到底物上)。具有反式 HNNH、与 Fe4 或 Fe6 结合或顺式 HNNH 与 Fe6 结合的结构也相当稳定。最后,在 TPSS 函数中,具有 cis-HNNH 侧接结合到簇的 Fe3-Fe4-Fe5-Fe7 面的结构也具有相当低的能量,但所有侧接结构均强烈受到 B3LYP 方法的不利影响。